- Latest available (Revised)
- Original (As adopted by EU)
Commission Regulation (EC) No 761/2009Show full title
Commission Regulation (EC) No 761/2009 of 23 July 2009 amending, for the purpose of its adaptation to technical progress, Regulation (EC) No 440/2008 laying down test methods pursuant to Regulation (EC) No 1907/2006 of the European Parliament and of the Council on the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH) (Text with EEA relevance)
You are here:

What Version
More Resources
Legislation originating from the EU
When the UK left the EU, legislation.gov.uk published EU legislation that had been published by the EU up to IP completion day (31 December 2020 11.00 p.m.). On legislation.gov.uk, these items of legislation are kept up-to-date with any amendments made by the UK since then.


This item of legislation originated from the EU
Legislation.gov.uk publishes the UK version. EUR-Lex publishes the EU version. The EU Exit Web Archive holds a snapshot of EUR-Lex’s version from IP completion day (31 December 2020 11.00 p.m.).
Status:
This is the original version as it was originally adopted in the EU.
This legislation may since have been updated - see the latest available (revised) version
ANNEX IA.4. VAPOUR PRESSURE
1.METHOD
This method is equivalent to OECD TG 104 (2004).
1.1.INTRODUCTION
This revised version of method A.4(1) includes one additional method; Effusion method: isothermal thermogravimetry, designed for substances with very low pressures (down to 10–10 Pa). In the light of needs for procedures, especially in relation to obtaining vapour pressure for substances with low vapour pressure, other procedures of this method are re-evaluated with respect to other applicability ranges.
At the thermodynamic equilibrium the vapour pressure of a pure substance is a function of temperature only. The fundamental principles are described elsewhere (2)(3).
No single measurement procedure is applicable to the entire range of vapour pressures from less than 10–10 to 105 Pa. Eight methods for measuring vapour pressure are included in this method which can be applied in different vapour pressure ranges. The various methods are compared as to application and measuring range in Table 1. The methods can only be applied for compounds that do not decompose under the conditions of the test. In cases where the experimental methods cannot be applied due to technical reasons, the vapour pressure can also be estimated, and a recommended estimation method is set out in the Appendix.
1.2.DEFINITIONS AND UNITS
The vapour pressure of a substance is defined as the saturation pressure above a solid or liquid substance.
The SI unit of pressure, which is the pascal (Pa), should be used. Other units which have been employed historically are given hereafter, together with their conversion factors:
| 1 Torr | = | 1 mm Hg | = | 1,333 × 102 Pa |
| 1 atmosphere | = | 1,013 × 105 Pa | ||
| 1 bar | = | 105 Pa |
The SI unit of temperature is the kelvin (K). The conversion of degrees Celsius to kelvin is according to the formula:
T = t + 273,15
where, T is the kelvin or thermodynamic temperature and t is the Celsius temperature.
Table 1
| a When using a capacitance manometer | |||||
| Measuring method | Substances | Estimated repeatability | Estimated reproducibility | Recommended range | |
|---|---|---|---|---|---|
| Solid | Liquid | ||||
| Dynamic method | Low melting | Yes | up to 25 % 1 to 5 % | up to 25 % 1 to 5 % | 103 Pa to 2 × 103 Pa 2 × 103 Pa to 105 Pa |
| Static method | Yes | Yes | 5 to 10 % | 5 to 10 % | 10 Pa to 105 Pa 10–2 Pa to 105 Paa |
| Isoteniscope method | Yes | Yes | 5 to 10 % | 5 to 10 % | 102 Pa to 105 Pa |
| Effusion method: vapour pressure balance | Yes | Yes | 5 to 20 % | up to 50 % | 10–3 to 1 Pa |
| Effusion method: Knudsen cell | Yes | Yes | 10 to 30 % | — | 10–10 to 1 P |
| Effusion method: isothermal thermogravimetry | Yes | Yes | 5 to 30 % | up to 50 % | 10–10 to 1 Pa |
| Gas saturation method | Yes | Yes | 10 to 30 % | up to 50 % | 10–10 to 103 Pa |
| Spinning rotor method | Yes | Yes | 10 to 20 % | — | 10–4 to 0,5 Pa |
1.3.PRINCIPLE OF THE TEST
In general, the vapour pressure is determined at various temperatures. In a limited temperature range, the logarithm of the vapour pressure of a pure substance is a linear function of the inverse of the thermodynamic temperature according to the simplified Clapeyron-Clausius equation:

where:
p
=
the vapour pressure in pascals
ΔHv
=
the heat of vaporisation in J mol–1
R
=
the universal gas constant, 8,314 J mol–1 K–1
T
=
the temperature in K
1.4.REFERENCE SUBSTANCES
Reference substances do not need to be employed. They serve primarily to check the performance of a method from time to time as well as to allow comparison between results of different methods.
1.5.DESCRIPTION OF THE METHOD
1.5.1.Dynamic method (Cottrell’s method)
1.5.1.1.Principle
The vapour pressure is determined by measuring the boiling temperature of the substance at various specified pressures between roughly 103 and 105 Pa. This method is also recommended for the determination of the boiling temperature. For that purpose it is useful up to 600 K. The boiling temperatures of liquids are approximately 0,1 °C higher at a depth of 3 to 4 cm than at the surface because of the hydrostatic pressure of the column of liquid. In Cottrell’s method (4) the thermometer is placed in the vapour above the surface of the liquid and the boiling liquid is made to pump itself continuously over the bulb of the thermometer. A thin layer of liquid which is in equilibrium with vapour at atmospheric pressure covers the bulb. The thermometer thus reads the true boiling point, without errors due to superheating or hydrostatic pressure. The pump originally employed by Cottrell is shown in figure 1. Tube A contains the boiling liquid. A platinum wire B sealed into the bottom facilitates uniform boiling. The side tube C leads to a condenser, and the sheath D prevents the cold condensate from reaching the thermometer E. When the liquid in A is boiling, bubbles and liquid trapped by the funnel are poured via the two arms of the pump F over the bulb of the thermometer.
| Figure 1  | Figure 2  |
Cottrell pump (4)
A:Thermocouple
B:Vacuum buffer volume
C:Pressure gauge
D:Vacuum
E:Measuring point
F:Heating element c.a. 150 W
1.5.1.2.Apparatus
A very accurate apparatus, employing the Cottrell principle, is shown in figure 2. It consists of a tube with a boiling section in the lower part, a cooler in the middle part, and an outlet and flange in the upper part. The Cottrell pump is placed in the boiling section which is heated by means of an electrical cartridge. The temperature is measured by a jacketed thermocouple, or resistance thermometer inserting through the flange at the top. The outlet is connected to the pressure regulation system. The latter consists of a vacuum pump, a buffer volume, a manostat for admitting nitrogen for pressure regulation and manometer.
1.5.1.3.Procedure
The substance is placed in the boiling section. Problems may be encountered with non-powder solids but these can sometimes be solved by heating the cooling jacket. The apparatus is sealed at the flange and the substance degassed. Frothing substances cannot be measured using this method.
The lowest desired pressure is then set and the heating is switched on. At the same time, the temperature sensor is connected to a recorder.
Equilibrium is reached when a constant boiling temperature is recorded at constant pressure. Particular care must be taken to avoid bumping during boiling. In addition, complete condensation must occur on the cooler. When determining the vapour pressure of low melting solids, care should be taken to prevent the condenser from blocking.
After recording this equilibrium point, a higher pressure is set. The process is continued in this manner until 105 Pa has been reached (approximately 5 to 10 measuring points in all). As a check, equilibrium points must be repeated at decreasing pressures.
1.5.2.Static method
1.5.2.1.Principle
In the static method (5), the vapour pressure at thermodynamic equilibrium is determined at a specified temperature. This method is suitable for substances and multicomponent liquids and solids in the range from 10–1 to 105 Pa and, provided care is taken, also in the range 1 to 10 Pa.
1.5.2.2.Apparatus
The equipment consists of a constant-temperature bath (precision of ±0,2 K), a container for the sample connected to a vacuum line, a manometer and a system to regulate the pressure. The sample chamber (figure 3a) is connected to the vacuum line via a valve and a differential manometer (U-tube containing a suitable manometer fluid) which serves as zero indicator. Mercury, silicones and phthalates are suitable for use in the differential manometer, depending on the pressure range and the chemical behaviour of the test substance. However, based on environmental concerns, the use of mercury should be avoided, if possible. The test substance must not dissolve noticeably in, or react with, the U-tube fluid. A pressure gauge can be used instead of a U-tube (figure 3b). For the manometer, mercury can be used in the range from normal pressure down to 102 Pa, while silicone fluids and phthalates are suitable for use below 102 Pa down to 10 Pa. There are other pressure gauges which can be used below 102 Pa and heatable membrane capacity manometers can even be used at below 10–1 Pa. The temperature is measured on the outside wall of the vessel containing the sample or in the vessel itself.
1.5.2.3.Procedure
Using the apparatus as described in figure 3a, fill the U-tube with the chosen liquid, which must be degassed at an elevated temperature before readings are taken. The test substance is placed in the apparatus and degassed at reduced temperature. In the case of a multiple-component sample, the temperature should be low enough to ensure that the composition of the material is not altered. Equilibrium can be established more quickly by stirring. The sample can be cooled with liquid nitrogen or dry ice, but care should be taken to avoid condensation of air or pump-fluid. With the valve over the sample vessel open, suction is applied for several minutes to remove the air. If necessary, the degassing operation is repeated several times.
| Figure 3a  | Figure 3b  |
When the sample is heated with the valve closed, the vapour pressure increases. This alters the equilibrium of the fluid in the U-tube. To compensate for this, nitrogen or air is admitted to the apparatus until the differential pressure indicator is at zero again. The pressure required for this can be read off the manometer or off an instrument of higher precision. This pressure corresponds to the vapour pressure of the substance at the temperature of the measurement. Using the apparatus described in figure 3b, the vapour pressure is read off directly.
The vapour pressure is determined at suitably small temperature intervals (approximately 5 to 10 measuring points in all) up to the desired temperature maximum.
Low-temperature readings must be repeated as a check. If the values obtained from the repeated readings do not coincide with the curve obtained for increasing temperature, this may be due to one of the following situations:
(i)
the sample still contains air (e.g. in the case of highly viscous materials) or low-boiling substances which is or are released during heating;
(ii)
the substance undergoes a chemical reaction in the temperature range investigated (e.g. decomposition, polymerisation).
1.5.3.Isoteniscope Method
1.5.3.1.Principle
The isoteniscope (6) is based on the principle of the static method. The method involves placing a sample in a bulb maintained at constant temperature and connected to a manometer and a vacuum pump. Impurities more volatile than the substance are removed by degassing at reduced pressure. The vapour pressure of the sample at selected temperatures is balanced by a known pressure of inert gas. The isoteniscope was developed to measure the vapour pressure of certain liquid hydrocarbons but it is appropriate for the investigation of solids as well. The method is usually not suitable for multicomponent systems. Results are subject to only slight errors for samples containing non-volatile impurities. The recommended range is 102 to 105 Pa.
1.5.3.2.Apparatus
An example of a measuring device is shown in figure 4. A complete description can be found in ASTM D 2879-86 (6).
1.5.3.3.Procedure
In the case of liquids, the substance itself serves as the fluid in the differential manometer. A quantity of the liquid, sufficient to fill the bulb and the short leg of the manometer, is put in the isoteniscope. The isoteniscope is attached to a vacuum system and evacuated, then filled by nitrogen. The evacuation and purge of the system is repeated twice to remove residual oxygen. The filled isoteniscope is placed in a horizontal position so that the sample spreads out into a thin layer in the sample bulb and manometer. The pressure of the system is reduced to 133 Pa and the sample is gently warmed until it just boils (removal of dissolved gases). The isoteniscope is then placed so that the sample returns to the bulb and fills the short leg of the manometer. The pressure is maintained at 133 Pa. The drawn-out tip of the sample bulb is heated with a small flame until the sample vapour released expands sufficiently to displace part of the sample from the upper part of the bulb and manometer arm into the manometer, creating a vapour-filled, nitrogen-free space. The isoteniscope is then placed in a constant temperature bath, and the pressure of the nitrogen is adjusted until it equals that of the sample. At the equilibrium, the pressure of the nitrogen equals the vapour pressure of the substance.

In the case of solids, and depending on the pressure and temperature ranges, manometer liquids such as silicon fluids or phthalates are used. The degassed manometer liquid is put in a bulge provided on the long arm of the isoteniscope. Then the solid to be investigated is placed in the sample bulb and is degassed at an elevated temperature. After that, the isoteniscope is inclined so that the manometer liquid can flow into the U-tube.
1.5.4.Effusion method: vapour pressure balance (7)
1.5.4.1.Principle
A sample of the test substance is heated in a small furnace and placed in an evacuated bell jar. The furnace is covered by a lid which carries small holes of known diameters. The vapour of the substance, escaping through one of the holes, is directed onto a balance pan of a highly sensitive balance which is also enclosed in the evacuated bell jar. In some designs the balance pan is surrounded by a refrigeration box, providing heat dissipation to the outside by thermal conduction, and is cooled by radiation so that the escaping vapour condenses on it. The momentum of the vapour jet acts as a force on the balance. The vapour pressure can be derived in two ways: directly from the force on the balance pan and also from the evaporation rate using the Hertz-Knudsen equation (2):

where:
G
=
evaporation rate (kg s–1 m–2)
M
=
molar mass (g mol–1)
T
=
temperature (K)
R
=
universal gas constant (J mol–1 K–1)
P
=
vapour pressure (Pa)
The recommended range is 10–3 to 1 Pa.
1.5.4.2.Apparatus
The general principle of the apparatus is illustrated in figure 5.

| A: | Base plate | F: | Refrigeration box and cooling bar |
| B: | Moving coil instrument | G: | Evaporator furnace |
| C: | Bell jar | H: | Dewar flask with liquid nitrogen |
| D: | Balance with scale pan | I: | Measurement of temperature of sample |
| E: | Vacuum measuring device | J: | Test Substance |
1.5.5.Effusion method: Knudsen cell
1.5.5.1.Principle
The method is based on the estimation of the mass of test substance flowing out per unit of time of a Knudsen cell (8) in the form of vapour, through a micro-orifice under ultra-vacuum conditions. The mass of effused vapour can be obtained either by determining the loss of mass of the cell or by condensing the vapour at low temperature and determining the amount of volatilised substance using chromatography. The vapour pressure is calculated by applying the Hertz-Knudsen relation (see section 1.5.4.1) with correction factors that depend on parameters of the apparatus (9). The recommended range is 10–10 to 1 Pa (10)(11)(12)(13)(14).
1.5.5.2.Apparatus
The general principle of the apparatus is illustrated in figure 6.

| 1: | Connection to vacuum | 7: | Threaded lid |
| 2: | Wells from platinum resistance thermometer or temperature measurement and control | 8: | Butterfly nuts |
| 3: | Lid for vacuum tank | 9: | Bolts |
| 4: | O-ring | 10: | Stainless steel effusion cells |
| 5: | Aluminum vacuum tank | 11: | Heater cartridge |
| 6: | Device for installing and removing the effusion cells |
1.5.6.Effusion method: isothermal thermogravimetry
1.5.6.1.Principle
The method is based on the determination of accelerated evaporation rates for the test substance at elevated temperatures and ambient pressure using thermogravimetry (10)(15)(16)(17)(18)(19)(20). The evaporation rates vT result from exposing the selected compound to a slowly flowing inert gas atmosphere, and monitoring the weight loss at defined isothermal temperatures T in Kelvin over appropriate periods of time. The vapour pressures pT are calculated from the vT values by using the linear relationship between the logarithm of the vapour pressure and the logarithm of the evaporation rate. If necessary, an extrapolation to temperatures of 20 and 25 °C can be made by regression analysis of log pT vs. 1/T. This method is suitable for substances with vapour pressures as low as 10–10 Pa (10–12 mbar) and with purity as close as possible to 100 % to avoid the misinterpretation of measured weight losses.
1.5.6.2.Apparatus
The general principle of the experimental set-up is shown in figure 7.
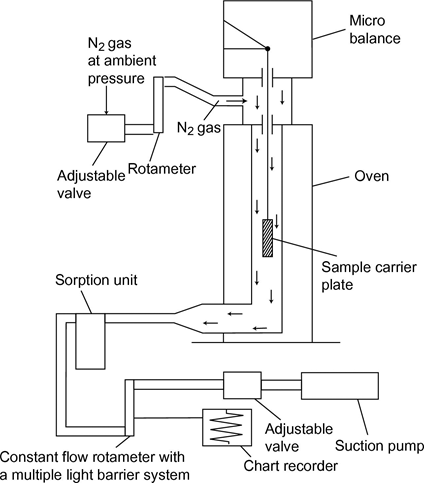
The sample carrier plate, hanging on a microbalance in a temperature controlled chamber, is swept by a stream of dry nitrogen gas which carries the vaporised molecules of the test substance away. After leaving the chamber, the gas stream is purified by a sorption unit.
1.5.6.3.Procedure
The test substance is applied to the surface of a roughened glass plate as a homogeneous layer. In the case of solids, the plate is wetted uniformly by a solution of the substance in a suitable solvent and dried in an inert atmosphere. For the measurement, the coated plate is hung into the thermogravimetric analyser and subsequently its weight loss is measured continuously as a function of time.
The evaporation rate vT at a definite temperature is calculated from the weight loss Δm of the sample plate by

where F is the surface area of the coated test substances, normally the surface area of the sample plate, and t is the time for weight loss Δm.
The vapour pressure pT is calculated on the basis of its function of evaporation rate vT:
Log pT = C + D · log vT
where C and D are constants specific for the experimental arrangement used, depending on the diameter of the measurement chamber and on the gas flow rate. These constants must be determined once by measuring a set of compounds with known vapour pressure and regressing log pT vs. log vT (11)(21)(22).
The relationship between the vapour pressure pT and the temperature T in Kelvin is given by
Log pT = A + B · 1/T
where A and B are constants obtained by regressing log pT vs. 1/T. With this equation, the vapour pressure can be calculated for any other temperature by extrapolation.
1.5.7.Gas saturation method (23)
1.5.7.1.Principle
Inert gas is passed, at room temperature and at a known flow rate, through or over a sample of the test substance, slowly enough to ensure saturation. Achieving saturation in the gas phase is of critical importance. The transported substance is trapped, generally using a sorbent, and its amount is determined. As an alternative to vapour trapping and subsequent analysis, in-train analytical techniques, like gas chromatography, may be used to determine quantitatively the amount of material transported. The vapour pressure is calculated on the assumption that the ideal gas law is obeyed and that the total pressure of a mixture of gases is equal to the sum of the pressures of the component gases. The partial pressure of the test substance, i.e. the vapour pressure, is calculated from the known total gas volume and from the weight of the material transported.
The gas saturation procedure is applicable to solid or liquid substances. It can be used for vapour pressures down to 10–10 Pa (10)(11)(12)(13)(14). The method is most reliable for vapour pressures below 103 Pa. Above 103 Pa, the vapour pressures are generally overestimated, probably due to aerosol formation. Since the vapour pressure measurements are made at room temperature, the need to extrapolate data from high temperatures is not necessary and high temperature extrapolation, which can often cause serious errors, is avoided.
1.5.7.2.Apparatus
The procedure requires the use of a constant-temperature box. The sketch in figure 8 shows a box containing three solid and three liquid sample holders, which allow for the triplicate analysis of either a solid or a liquid sample. The temperature is controlled to ±0,5 °C or better.

In general, nitrogen is used as an inert carrier gas but, occasionally, another gas may be required (24). The carrier gas must be dry. The gas stream is split into 6 streams, controlled by needle valves (approximately 0,79 mm orifice), and flows into the box via 3,8 mm i.d. copper tubing. After temperature equilibration, the gas flows through the sample and the sorbent trap and exists from the box.
Solid samples are loaded into 5 mm i.d. glass tubing between glass wool plugs (see Figure 9). Figure 10 shows a liquid sample holder and sorbent system. The most reproducible method for measuring the vapour pressure of liquids is to coat the liquid on glass beads or on an inert sorbent such as silica, and to pack the holder with these beads. As an alternative, the carrier gas may be made to pass a coarse frit and bubble through a column of the liquid test substance.
| Figure 9  | Figure 10  |
The sorbent system contains a front and a backup sorbent section. At very low vapour pressures, only small amounts are retained by the sorbent and the adsorption on the glass wool and the glass tubing between the sample and the sorbent may be a serious problem.
Traps cooled with solid CO2 are another efficient way for collecting the vaporised material. They do not cause any back pressure on the saturator column and it is also easy to quantitatively remove the trapped material.
1.5.7.3.Procedure
The flow rate of the effluent carrier gas is measured at room temperature. The flow rate is checked frequently during the experiment to assure that there is an accurate value for the total volume of carrier gas. Continuous monitoring with a mass flow-meter is preferred. Saturation of the gas phase may require considerable contact time and hence quite low gas flow rates (25).
At the end of the experiment, both the front and backup sorbent sections are analysed separately. The compound on each section is desorbed by adding a solvent. The resulting solutions are analysed quantitatively to determine the weight desorbed from each section. The choice of the analytical method (also the choice of sorbent and desorbing solvent) is dictated by the nature of the test material. The desorption efficiency is determined by injecting a known amount of sample onto the sorbent, desorbing it and analysing the amount recovered. It is important to check the desorption efficiency at or near the concentration of the sample under the test conditions.
To assure that the carrier gas is saturated with the test substance, three different gas flow rates are used. If the calculated vapour pressure shows no dependence on flow rate, the gas is assumed to be saturated.
The vapour pressure is calculated through the equation:

where:
p
=
vapour pressure (Pa)
W
=
mass of evaporated test substance (g)
V
=
volume of saturated gas (m3)
R
=
universal gas constant 8,314 (J mol–1 K–1)
T
=
temperature (K)
M
=
molar mass of test substance (g mol–1)
Measured volumes must be corrected for pressure and temperature differences between the flow meter and the saturator.
1.5.8.Spinning rotor
1.5.8.1.Principle
This method uses a spinning rotor viscosity gauge, in which the measuring element is a small steel ball which, suspended in a magnetic field, is made to spin by rotating fields (26)(27)(28). Pick-up coils allow its spinning rate to be measured. When the ball has reached a given rotational speed, usually about 400 revolutions per second, energising is stopped and deceleration, due to gas friction, takes place. The drop of rotational speed is measured as a function of time. The vapour pressure is deduced from the pressure-dependent slow-down of the steel ball. The recommended range is 10–4 to 0,5 Pa.
1.5.8.2.Apparatus
A schematic drawing of the experimental set-up is shown in figure 11. The measuring head is placed in a constant-temperature enclosure, regulated within 0,1 °C. The sample container is placed in a separate enclosure, also regulated within 0,1 °C. All other parts of the set-up are kept at a higher temperature to prevent condensation. The whole apparatus is connected to a high-vacuum system.

2.DATA AND REPORTING
2.1.DATA
The vapour pressure from any of the preceding methods should be determined for at least two temperatures. Three or more are preferred in the range from 0 to 50 °C, in order to check the linearity of the vapour pressure curve. In case of Effusion method (Knudsen cell and isothermal thermogravimetry) and Gas saturation method, 120 to 150 °C is recommended for the measuring temperature range instead of 0 to 50 °C.
2.2.TEST REPORT
The test report must include the following information:
method used,
precise specification of the substance (identity and impurities) and preliminary purification step, if any,
at least two vapour pressure and temperature values — and preferably three or more — required in the range from 0 to 50 °C (or 120 to 150 °C),
at least one of the temperatures should be at or below 25 °C, if technically possible according to the chosen method,
all original data,
a log p versus 1/T curve,
an estimate of the vapour pressure at 20 or 25 °C.
If a transition (change of state, decomposition) is observed, the following information should be noted:
nature of the change,
temperature at which the change occurs at atmospheric pressure,
vapour pressure at 10 and 20 °C below the transition temperature and 10 and 20 °C above this temperature (unless the transition is from solid to gas).
All information and remarks relevant for the interpretation of results have to be reported, especially with regard to impurities and physical state of the substance.
3.LITERATURE
(1)
Official Journal of the European Communities L 383 A, 26-47 (1992).
(2)
Ambrose, D. (1975). Experimental Thermodynamics, Vol. II, Le Neindre, B., and Vodar, B., Eds., Butterworths, London.
(3)
Weissberger R., ed. (1959). Technique of Organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed., Vol. I, Part I. Chapter IX, Interscience Publ., New York.
(4)
Glasstone, S. (1946). Textbook of Physical Chemistry, 2nd ed., Van Nostrand Company, New York.
(5)
NF T 20-048 AFNOR (September 1985). Chemical products for industrial use — Determination of vapour pressure of solids and liquids within a range from 10–1 to 105 Pa — Static method.
(6)
ASTM D 2879-86, Standard test method for vapour pressure — temperature relationship and initial decomposition temperature of liquids by isoteniscope.
(7)
NF T 20-047 AFNOR (September 1985). Chemical products for industrial use — Determination of vapour pressure of solids and liquids within range from 10–3 to 1 Pa — Vapour pressure balance method.
(8)
Knudsen, M. (1909). Ann. Phys. Lpz., 29, 1979; (1911), 34, 593.
(9)
Ambrose, D., Lawrenson, I.J., Sprake, C.H.S. (1975). J. Chem. Thermodynamics 7, 1173.
(10)
Schmuckler, M.E., Barefoot, A.C., Kleier, D.A., Cobranchi, D.P. (2000), Vapor pressures of sulfonylurea herbicides; Pest Management Science 56, 521-532.
(11)
Tomlin, C.D.S. (ed.), The Pesticide Manual, Twelfth Edition (2000).
(12)
Friedrich, K., Stammbach, K., Gas chromatographic determination of small vapour pressures determination of the vapour pressures of some triazine herbicides. J. Chromatog. 16 (1964), 22-28.
(13)
Grayson, B.T., Fosbraey, L.A., Pesticide Science 16 (1982), 269-278.
(14)
Rordorf, B.F., Prediction of vapor pressures, boiling points and enthalpies of fusion for twenty-nine halogenated dibenzo-p-dioxins, Thermochimia Acta 112 Issue 1 (1987), 117-122.
(15)
Gückel, W., Synnatschke, G., Ritttig, R., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection; Pesticide Science 4 (1973) 137-147.
(16)
Gückel, W., Synnatschke, G., Ritttig, R., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection II. Application to Formulated Products; Pesticide Science 5 (1974) 393-400.
(17)
Gückel, W., Kaestel, R., Lewerenz, J., Synnatschke, G., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection. Part III: The Temperature Relationship between Vapour Pressure and Evaporation Rate; Pesticide Science 13 (1982) 161-168.
(18)
Gückel, W., Kaestel, R., Kroehl, T., Parg, A., Methods for Determining the Vapour Pressure of Active Ingredients Used in Crop Protection. Part IV: An Improved Thermogravimetric Determination Based on Evaporation Rate; Pesticide Science 45 (1995) 27-31.
(19)
Kroehl, T., Kaestel, R., Koenig, W., Ziegler, H., Koehle, H., Parg, A., Methods for Determining the Vapour Pressure of Active Ingredients Used in Crop Protection. Part V: Thermogravimetry Combined with Solid Phase MicroExtraction (SPME); Pesticide Science, 53 (1998) 300-310.
(20)
Tesconi, M., Yalkowsky, S.H., A Novel Thermogravimetric Method for Estimating the Saturated Vapor Pressure of Low-Volatility Compounds; Journal of Pharmaceutical Science 87(12) (1998) 1512-20.
(21)
Lide, D.R. (ed.), CRC Handbook of Chemistry and Physics, 81st ed. (2000), Vapour Pressure in the Range — 25 °C to 150 °C.
(22)
Meister, R.T. (ed.), Farm Chemicals Handbook, Vol. 88 (2002).
(23)
40 CFR, 796. (1993). pp 148-153, Office of the Federal Register, Washington DC.
(24)
Rordorf B.F. (1985). Thermochimica Acta 85, 435.
(25)
Westcott et al. (1981). Environ. Sci. Technol. 15, 1375.
(26)
Messer G., Röhl, P., Grosse G., and Jitschin W. (1987). J. Vac. Sci. Technol. (A), 5(4), 2440.
(27)
Comsa G., Fremerey J.K., and Lindenau, B. (1980). J. Vac. Sci. Technol. 17(2), 642.
(28)
Fremerey, J.K. (1985). J. Vac. Sci. Technol. (A), 3(3), 1715.
AppendixEstimation method
INTRODUCTION
Estimated values of the vapour pressure can be used:
for deciding which of the experimental methods is appropriate,
for providing an estimate or limit value in cases where the experimental method cannot be applied due to technical reasons.
ESTIMATION METHOD
The vapour pressure of liquids and solids can be estimated by use of the modified Watson correlation (a). The only experimental data required is the normal boiling point. The method is applicable over the pressure range from 105 Pa to 10–5 Pa.
Detailed information on the method is given in ‘Handbook of Chemical Property Estimation Methods’ (b). See also OECD Environmental Monograph No.67 (c).
CALCULATION PROCEDURE
The vapour pressure is calculated as follows:

where:
T
=
temperature of interest
Tb
=
normal boiling point
Pvp
=
vapour pressure at temperature T
ΔHvb
=
heat of vaporisation
ΔZb
=
compressibility factor (estimated at 0,97)
m
=
empirical factor depending on the physical state at the temperature of interest
Further,

where, KF is an empirical factor considering the polarity of the substance. For several compound types, KF factors are listed in reference (b).
Quite often, data are available in which a boiling point at reduced pressure is given. In such a case, the vapour pressure is calculated as follows:

where, T1 is the boiling point at the reduced pressure P1.
REPORT
When using the estimation method, the report shall include a comprehensive documentation of the calculation.
LITERATURE
(a)
Watson, K.M. (1943). Ind. Eng. Chem, 35, 398.
(b)
Lyman, W.J., Reehl, W.F., Rosenblatt, D.H. (1982). Handbook of Chemical Property Estimation Methods, McGraw-Hill.
(c)
OECD Environmental Monograph No.67. Application of Structure-Activity Relationships to the Estimation of Properties Important in Exposure Assessment (1993).
Options/Help
Print Options
PrintThe Whole Regulation
PrintThis Annex only
You have chosen to open the Whole Regulation
The Whole Regulation you have selected contains over 200 provisions and might take some time to download. You may also experience some issues with your browser, such as an alert box that a script is taking a long time to run.
Would you like to continue?
You have chosen to open Schedules only
The Schedules you have selected contains over 200 provisions and might take some time to download. You may also experience some issues with your browser, such as an alert box that a script is taking a long time to run.
Would you like to continue?
Legislation is available in different versions:
Latest Available (revised):The latest available updated version of the legislation incorporating changes made by subsequent legislation and applied by our editorial team. Changes we have not yet applied to the text, can be found in the ‘Changes to Legislation’ area.
Original (As adopted by EU): The original version of the legislation as it stood when it was first adopted in the EU. No changes have been applied to the text.
Opening Options
Different options to open legislation in order to view more content on screen at once
More Resources
Access essential accompanying documents and information for this legislation item from this tab. Dependent on the legislation item being viewed this may include:
- the original print PDF of the as adopted version that was used for the EU Official Journal
- lists of changes made by and/or affecting this legislation item
- all formats of all associated documents
- correction slips
- links to related legislation and further information resources
More Resources
Use this menu to access essential accompanying documents and information for this legislation item. Dependent on the legislation item being viewed this may include:
- the original print PDF of the as adopted version that was used for the print copy
- correction slips
Click 'View More' or select 'More Resources' tab for additional information including:
- lists of changes made by and/or affecting this legislation item
- confers power and blanket amendment details
- all formats of all associated documents
- links to related legislation and further information resources
 All content is available under the Open Government Licence v3.0 except where otherwise stated. This site additionally contains content derived from EUR-Lex, reused under the terms of the Commission Decision 2011/833/EU on the reuse of documents from the EU institutions. For more information see the EUR-Lex public statement on re-use.
All content is available under the Open Government Licence v3.0 except where otherwise stated. This site additionally contains content derived from EUR-Lex, reused under the terms of the Commission Decision 2011/833/EU on the reuse of documents from the EU institutions. For more information see the EUR-Lex public statement on re-use.