- Latest available (Revised)
- Point in Time (15/02/2018)
- Original (As adopted by EU)
Regulation (EU) No 1007/2011 of the European Parliament and of the CouncilShow full title
Regulation (EU) No 1007/2011 of the European Parliament and of the Council of 27 September 2011 on textile fibre names and related labelling and marking of the fibre composition of textile products and repealing Council Directive 73/44/EEC and Directives 96/73/EC and 2008/121/EC of the European Parliament and of the Council (Text with EEA relevance)
You are here:
- Regulations originating from the EU
- 2011 No. 1007
- Annexes only

What Version
Advanced Features
- Show Geographical Extent(e.g. England, Wales, Scotland and Northern Ireland)
- Show Timeline of Changes
Opening Options
More Resources
Revised version PDFs
- Revised 15/02/20180.78 MB
- Revised 01/07/20130.73 MB
- Revised 08/05/20120.72 MB
- Revised 21/12/20110.71 MB
This is a Regulation originating from the EU


This is a legislation item that originated from the EU
After exit day there will be three versions of this legislation to consult for different purposes. The legislation.gov.uk version is the version that applies in the UK. The EU Version currently on EUR-lex is the version that currently applies in the EU i.e you may need this if you operate a business in the EU.
The web archive version is the official version of this legislation item as it stood on exit day before being published to legislation.gov.uk and any subsequent UK changes and effects applied. The web archive also captured associated case law and other language formats from EUR-Lex.
Changes over time for: Regulation (EU) No 1007/2011 of the European Parliament and of the Council (Annexes only)
Status:
Point in time view as at 15/02/2018.
Changes to legislation:
There are outstanding changes not yet made to Regulation (EU) No 1007/2011 of the European Parliament and of the Council. Any changes that have already been made to the legislation appear in the content and are referenced with annotations.
Changes to Legislation
Changes and effects yet to be applied by the editorial team are only applicable when viewing the latest version or prospective version of legislation. They are therefore not accessible when viewing legislation as at a specific point in time. To view the ‘Changes to Legislation’ information for this provision return to the latest version view using the options provided in the ‘What Version’ box above.
ANNEX IU.K.
List of textile fibre names
(referred to in Article 5)
| Table 1 | ||
| Number | Name | Fibre description |
|---|---|---|
| 1 | wool | fibre from sheep's or lambs’ fleeces (Ovis aries) or a mixture of fibres from sheep's or lambs’ fleeces and the hairs of animals listed in number 2 |
| 2 | alpaca, llama, camel, cashmere, mohair, angora, vicuna, yak, guanaco, cashgora, beaver, otter, followed or not by the word ‘wool’ or ‘hair’ | hair of the following animals: alpaca, llama, camel, kashmir goat, angora goat, angora rabbit, vicuna, yak, guanaco, cashgora goat, beaver, otter |
| 3 | animal or horsehair, with or without an indication of the kind of animal (e.g. cattle hair, common goat hair, horsehair) | hair of the various animals not mentioned under number 1 or 2 |
| 4 | Silk | fibre obtained exclusively from silk-secreting insects |
| 5 | cotton | fibre obtained from the bolls of the cotton plant (Gossypium) |
| 6 | kapok | fibre obtained from the inside of the kapok fruit (Ceiba pentandra) |
| 7 | flax (or linen) | fibre obtained from the bast of the flax plant (Linum usitatissimum) |
| 8 | true hemp | fibre obtained from the bast of hemp (Cannabis sativa) |
| 9 | Jute | fibre obtained from the bast of Corchorus olitorius and Corchorus capsularis. For the purposes of this Regulation, bast fibres obtained from the following species shall be treated in the same way as jute: Hibiscus cannabinus, Hibiscus sabdariffa, Abutilon avicennae, Urena lobata, Urena sinuata |
| 10 | abaca (Manila hemp) | fibre obtained from the sheathing leaf of Musa textilis |
| 11 | Alfa | fibre obtained from the leaves of Stipa tenacissima |
| 12 | coir (coconut) | fibre obtained from the fruit of Cocos nucifera |
| 13 | broom | fibre obtained from the bast of Cytisus scoparius and/or Spartium Junceum |
| 14 | ramie | fibre obtained from the bast of Boehmeria nivea and Boehmeria tenacissima |
| 15 | sisal | fibre obtained from the leaves of Agave sisalana |
| 16 | sunn | fibre from the bast of Crotalaria juncea |
| 17 | henequen | fibre from the bast of Agave fourcroydes |
| 18 | maguey | fibre from the bast of Agave cantala |
| Table 2 | ||
| Number | Name | Fibre description |
|---|---|---|
| 19 | acetate | cellulose acetate fibre wherein less than 92 % but at least 74 % of the hydroxyl groups are acetylated |
| 20 | alginate | fibre obtained from metallic salts of alginic acid |
| 21 | cupro | regenerated cellulose fibre obtained by the cuprammonium process |
| 22 | modal | a regenerated cellulose fibre obtained by a modified viscose process having a high breaking force and high wet modulus. The breaking force (BC) in the conditioned state and the force (BM) required to produce an elongation of 5 % in the wet state are: where T is the mean linear density in decitex |
| 23 | protein | fibre obtained from natural protein substances regenerated and stabilised through the action of chemical agents |
| 24 | triacetate | cellulose acetate fibre wherein at least 92 % of the hydroxyl groups are acetylated |
| 25 | viscose | regenerated cellulose fibre obtained by the viscose process for filament and discontinuous fibre |
| 26 | acrylic | fibre formed of linear macromolecules comprising at least 85 % (by mass) in the chain of the acrylonitrilic pattern |
| 27 | chlorofibre | fibre formed of linear macromolecules having in their chain more than 50 % by mass of chlorinated vinyl or chlorinated vinylidene monomeric units |
| 28 | fluorofibre | fibre formed of linear macromolecules made from fluorocarbon aliphatic monomers |
| 29 | modacrylic | fibre formed of linear macromolecules having in the chain more than 50 % and less than 85 % (by mass) of the acrylonitrilic pattern |
| 30 | polyamide or nylon | fibre formed from synthetic linear macromolecules having in the chain recurring amide linkages of which at least 85 % are joined to aliphatic or cycloaliphatic units |
| 31 | aramid | fibre formed from synthetic linear macromolecules made up of aromatic groups joined by amide or imide linkages, of which at least 85 % are joined directly to two aromatic rings and with the number of imide linkages, if present, not exceeding the number of amide linkages |
| 32 | polyimide | fibre formed from synthetic linear macromolecules having in the chain recurring imide units |
| 33 | lyocell | a regenerated cellulose fibre obtained by dissolution, and an organic solvent (mixture of organic chemicals and water) spinning process, without formation of derivatives |
| 34 | polylactide | fibre formed of linear macromolecules having in the chain at least 85 % (by mass) of lactic acid ester units derived from naturally occurring sugars, and which has a melting temperature of at least 135 °C |
| 35 | polyester | fibre formed of linear macromolecules comprising at least 85 % (by mass) in the chain of an ester of a diol and terephthalic acid |
| 36 | polyethylene | fibre formed of un-substituted aliphatic saturated hydrocarbon linear macromolecules |
| 37 | polypropylene | fibre formed of an aliphatic saturated hydrocarbon linear macromolecule where one carbon atom in two carries a methyl side chain in an isotactic disposition and without further substitution |
| 38 | polycarbamide | fibre formed of linear macromolecules having in the chain the recurring ureylene (NH-CO-NH) functional group |
| 39 | polyurethane | fibre formed of linear macromolecules composed of chains with the recurring urethane functional group |
| 40 | vinylal | fibre formed of linear macromolecules whose chain is constituted by poly(vinyl alcohol) with differing levels of acetalisation |
| 41 | trivinyl | fibre formed of acrylonitrile terpolymer, a chlorinated vinyl monomer and a third vinyl monomer, none of which represents as much as 50 % of the total mass |
| 42 | elastodiene | elastofibre composed of natural or synthetic polyisoprene, or composed of one or more dienes polymerised with or without one or more vinyl monomers, and which, when stretched to three times its original length and released, recovers rapidly and substantially to its initial length |
| 43 | elastane | elastofibre composed of at least 85 % (by mass) of a segmented polyurethane, and which, when stretched to three times its original length and released, recovers rapidly and substantially to its initial length |
| 44 | glass fibre | fibre made of glass |
| 45 | elastomultiester | fibre formed by interaction of two or more chemically distinct linear macromolecules in two or more distinct phases (of which none exceeds 85 % by mass) which contains ester groups as the dominant functional unit (at least 85 %) and which, after suitable treatment when stretched to one and half times its original length and released, recovers rapidly and substantially to its initial length |
| 46 | elastolefin | fibre composed of at least 95 % (by mass) of macromolecules partially cross-linked, made up from ethylene and at least one other olefin and which, when stretched to one and a half times its original length and released, recovers rapidly and substantially to its initial length |
| 47 | melamine | fibre formed of at least 85 % by mass of cross-linked macromolecules made up of melamine derivatives |
| 48 | name corresponding to the material of which the fibres are composed, e.g. metal (metallic, metallised), asbestos, paper, followed or not by the word ‘yarn’ or ‘fibre’ | fibres obtained from miscellaneous or new materials not listed above |
| [F149 | polypropylene/polyamide bicomponent | a bicomponent fibre composed of between 10 % and 25 % by mass of polyamide fibrils embedded in polypropylene matrix] |
| [F250 | polyacrylate | fibre formed of cross-linked macromolecules having more than 35 % (by mass) of acrylate groups (acid, light metal salts or esters) and less than 10 % (by mass) of acrylonitrile groups in the chain and up to 15 % (by mass) of nitrogen in the cross-linking] |
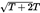
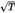
Textual Amendments
F1 Inserted by Commission Delegated Regulation (EU) No 286/2012 of 27 January 2012 amending, in order to include a new textile fibre name, Annex I, and, for the purposes of their adaptation to technical progress, Annexes VIII and IX to Regulation (EU) No 1007/2011 of the European Parliament and of the Council on textile fibre names and related labelling and marking of the fibre composition of textile products (Text with EEA relevance).
F2 Inserted by Commission Delegated Regulation (EU) 2018/122 of 20 October 2017 amending Annexes I, II, VI, VIII and IX to Regulation (EU) No 1007/2011 of the European Parliament and of the Council on textile fibre names and related labelling and marking of the fibre composition of textile products (Text with EEA relevance).
ANNEX IIU.K. Minimum requirements regarding a technical file to be included in the application for a new textile fibre name (referred to in Article 6)
A technical file to be attached to an application for the inclusion of a new textile fibre name in the list set out in Annex I, as provided for in Article 6, shall contain at least the following information:
(1)
Proposed name of the textile fibre:
The name proposed shall be related to the chemical composition and shall provide information about the characteristics of the fibre, if appropriate. The name proposed shall be free of any intellectual property rights and shall not be linked to the manufacturer.
(2)
[F3Proposed definition of the textile fibre:
The definition proposed shall describe the fibre composition. The characteristics mentioned in the definition of the new textile fibre, such as elasticity, shall be verifiable via standard test methods to be provided with the technical file along with the experimental results of analyses.
(3)
Identification of the textile fibre: chemical formula, differences from existing textile fibres, FTIR spectrum together with, where relevant, detailed data such as melting point, density, refractive index and burning behaviour.]
(4)
Proposed agreed allowance to be used in the calculation of fibre composition.
(5)
[F3Proposed identification and quantification methods, including experimental data:
The applicant shall evaluate the possibility to use the methods listed in Annex VIII or the harmonised standards to be introduced in that Annex to analyse the most expected commercial mixtures of the new textile fibre with other textile fibres and shall propose at least one of those methods. For those methods or harmonised standards where the textile fibre can be considered as an insoluble component, the applicant shall indicate the ‘ d ’ factors, which correspond to the mass correction factors to be applied for the calculations (to account for the loss in mass, known to occur during the analysis) of the new textile fibre.
If methods listed in this Regulation are not suitable, the applicant shall provide adequate reasoning and propose one or more new methods. The proposed new method or methods shall describe the field of application (including fibre mixtures), the principle (notably chemical process and steps), the apparatus and reagent or reagents, the test procedure, the calculation and expression of results (including the value of ‘ d ’ factors), and the precision (confidence limits of results).
The application shall contain all the experimental data, in particular regarding fibre characteristics, identification and quantification methods proposed. Data on the accuracy, robustness and repeatability of the methods shall be provided with the file.]
(6)
Available scientific information concerning possible allergic reactions or other adverse effects of the new textile fibre on human health, including results of tests conducted to that effect in compliance with relevant Union legislation.
(7)
[F3Additional information on production process and consumer relevance to support the application:
The technical file shall, at least, contain information on the number of producers, the location of production facilities and the expected market availability of the new fibre or of products manufactured from that fibre.]
(8)
[F2Availability of samples:
The manufacturer or any person acting on the manufacturer's behalf shall provide representative samples of the new pure textile fibre and the relevant textile fibre mixtures necessary for verifying the accuracy, robustness and repeatability of the proposed identification and quantification methods. The Commission may request additional samples of relevant fibre mixtures from the manufacturer or the person acting on the manufacturer's behalf.]
Textual Amendments
F3 Substituted by Commission Delegated Regulation (EU) 2018/122 of 20 October 2017 amending Annexes I, II, VI, VIII and IX to Regulation (EU) No 1007/2011 of the European Parliament and of the Council on textile fibre names and related labelling and marking of the fibre composition of textile products (Text with EEA relevance).
The manufacturer or any person acting on the manufacturer’s behalf shall provide representative samples of the new pure textile fibre and the relevant textile fibre mixtures necessary to conduct the validation of the proposed identification and quantification methods. The Commission may request additional samples of relevant fibre mixtures from the manufacturer or the person acting on the manufacturer’s behalf.
ANNEX IIIU.K. Names referred to in Article 8(1)
— in Bulgarian
:
‘необработена вълна’
— in Spanish
:
‘lana virgen’ or ‘lana de esquilado’
— in Czech
:
‘střižní vlna’
— in Danish
:
‘ren, ny uld’
— in German
:
‘Schurwolle’
— in Estonian
:
‘uus vill’
— in Greek
:
‘παρθένο μαλλί’
— in English
:
‘fleece wool’ or ‘virgin wool’
— in French
:
‘laine vierge’ or ‘laine de tonte’
— in Irish
:
‘olann lomra’
— in Italian
:
‘lana vergine’ or ‘lana di tosa’
— in Latvian
:
‘pirmlietojuma vilna’ or ‘cirptā vilna’
— in Lithuanian
:
‘natūralioji vilna’
— in Hungarian
:
‘élőgyapjú’
— in Maltese
:
‘suf verġni’
— in Dutch
:
‘scheerwol’
— in Polish
:
‘żywa wełna’
— in Portuguese
:
‘lã virgem’
— in Romanian
:
‘lână virgină’
— in Slovak
:
‘strižná vlna’
— in Slovene
:
‘runska volna’
— in Finnish
:
‘uusi villa’
— in Swedish
:
‘ny ull’
Textual Amendments
F4 Inserted by Council Regulation (EU) No 517/2013 of 13 May 2013 adapting certain regulations and decisions in the fields of free movement of goods, freedom of movement for persons, company law, competition policy, agriculture, food safety, veterinary and phytosanitary policy, transport policy, energy, taxation, statistics, trans-European networks, judiciary and fundamental rights, justice, freedom and security, environment, customs union, external relations, foreign, security and defence policy and institutions, by reason of the accession of the Republic of Croatia.
ANNEX IVU.K.
Special provisions for the labelling and marking of certain textile products
(referred to in Article 13)
ANNEX VU.K. Textile products for which labelling or marking is not mandatory (referred to in Article 17(2))
1.Sleeve-supporting armbandsU.K.
2.Watch straps of textile materialsU.K.
3.Labels and badgesU.K.
4.Stuffed pan-holders of textile materialsU.K.
5.Coffee cosy coversU.K.
6.Tea cosy coversU.K.
7.Sleeve protectorsU.K.
8.Muffs other than in pile fabricU.K.
9.Artificial flowersU.K.
10.Pin cushionsU.K.
11.Painted canvasU.K.
12.Textile products for base and underlying fabrics and stiffeningsU.K.
13.Old made-up textile products, where explicitly stated to be suchU.K.
14.GaitersU.K.
15.Packaging, not new and sold as suchU.K.
16.Fancy goods and saddlery, of textile materialsU.K.
17.Travel goods of textile materialsU.K.
18.Hand-embroidered tapestries, finished or unfinished, and materials for their production, including embroidery yarns, sold separately from the canvas and specially presented for use in such tapestriesU.K.
19.Slide fastenersU.K.
20.Buttons and buckles covered with textile materialsU.K.
21.Book covers of textile materialsU.K.
22.ToysU.K.
23.Textile parts of footwearU.K.
24.Table mats having several components and a surface area of not more than 500 cm2 U.K.
25.Oven gloves and clothsU.K.
26.Egg cosy coversU.K.
27.Make-up casesU.K.
28.Tobacco pouches of textile fabricU.K.
29.Spectacle, cigarette and cigar, lighter and comb cases of textile fabricU.K.
30.Covers for mobile telephones and portable media players with a surface of not more than 160 cm2 U.K.
31.Protective requisites for sports with the exception of glovesU.K.
32.Toilet casesU.K.
33.Shoe-cleaning casesU.K.
34.Funeral productsU.K.
35.Disposable products, with the exception of waddingU.K.
36.Textile products subject to the rules of the European Pharmacopoeia and covered by a reference to those rules, non-disposable bandages for medical and orthopaedic use and orthopaedic textile products in generalU.K.
37.Textile products including cordage, ropes and string, subject to item 12 of Annex VI, normally intended:U.K.
(a)
for use as equipment components in the manufacture and processing of goods;
(b)
for incorporation in machines, installations (e.g. for heating, air conditioning or lighting), domestic and other appliances, vehicles and other means of transport, or for their operation, maintenance or equipment, other than tarpaulin covers and textile motor vehicle accessories sold separately from the vehicle
38.Textile products for protection and safety purposes such as safety belts, parachutes, life-jackets, emergency chutes, fire-fighting devices, bulletproof waistcoats and special protective garments (e.g. protection against fire, chemical substances or other safety hazards)U.K.
39.Air-supported structures (e.g. sports halls, exhibition stands or storage facilities), provided that details of the performances and technical specifications of these products are suppliedU.K.
40.SailsU.K.
41.Animal clothingU.K.
42.Flags and bannersU.K.
ANNEX VIU.K. Textile products for which inclusive labelling is sufficient (referred to in Article 17(3))
1.FloorclothsU.K.
2.Cleaning clothsU.K.
3.Edgings and trimmingsU.K.
4.PassementerieU.K.
5.BeltsU.K.
6.BracesU.K.
7.Suspenders and gartersU.K.
8.Shoe and boot lacesU.K.
9.RibbonsU.K.
10.ElasticU.K.
11.New packaging sold as suchU.K.
12.Packing string and agricultural twine; string, cordage and ropes other than those falling within item 37 of Annex V(1) U.K.
13.Table matsU.K.
14.HandkerchiefsU.K.
15.Bun nets and hair netsU.K.
16.Ties and bow ties for childrenU.K.
17.Bibs, washgloves and face flannelsU.K.
[F318. Sewing, mending and embroidery yarns presented for retail sale] U.K.
19.Tape for curtains and blinds and shuttersU.K.
ANNEX VIIU.K.
Items not to be taken into account for the determination of fibre composition
(referred to in Article 19(2))
ANNEX VIIIU.K. Methods for the quantitative analysis of binary and ternary textile fibre mixtures (referred to in Article 19(1))
CHAPTER 1U.K.
I. Preparation of laboratory test samples and test specimens to determine the fibre composition of textile products U.K.
1.FIELD OF APPLICATIONU.K.
This Chapter gives procedures for obtaining laboratory test samples of a suitable size for pre-treatment for quantitative analysis (i.e. of a mass not exceeding 100 g) from laboratory bulk samples, and for selecting test specimens from the laboratory test samples that have been pre-treated to remove non-fibrous matter(2).
2.DEFINITIONSU.K.
2.1.Bulk sourceU.K.
The quantity of material which is assessed on the basis of one series of test results. This may comprise, for example, all the material in one delivery of cloth; all the cloth woven from a particular beam; a consignment of yarn, a bale or a group of bales of raw fibre.
2.2.Laboratory bulk sampleU.K.
The portion of the bulk source taken to be representative of the whole, and which is available to the laboratory. The size and nature of the laboratory bulk sample shall be sufficient to adequately overcome the variability of the bulk source and to facilitate ease of handling in the laboratory(3).
2.3.Laboratory test sampleU.K.
That portion of the laboratory bulk sample that is subjected to pre-treatment to remove non-fibrous matter, and from which test specimens are taken. The size and nature of the laboratory test sample shall be sufficient to overcome adequately the variability of the laboratory bulk sample(4).
2.4.Test specimenU.K.
The portion of material required to give an individual test result, and selected from the laboratory test sample.
3.PRINCIPLEU.K.
The laboratory test sample is selected so that it is representative of the laboratory bulk sample.
The test specimens are taken from the laboratory test sample in such a way that each of them is representative of the laboratory test sample.
4.SAMPLING FROM LOOSE FIBRESU.K.
4.1.Unorientated fibresU.K.
Obtain the laboratory test sample by selecting tufts at random from the laboratory bulk sample. Mix thoroughly the whole of the laboratory test sample by means of a laboratory carder(5). Subject the web or mixture, including loose fibres and fibres adhering to the equipment used for mixing, to pre-treatment. Then select test specimens, in proportion to the respective masses, from the web or mixture, from the loose fibres and from the fibres adhering to the equipment.
If the card web remains intact after pre-treatment, select the test specimens in the manner described in 4.2. If the card web is disturbed by the pre-treatment, select each test specimen by removing at random at least 16 small tufts of suitable and approximately equal size and then combine them.
4.2.Orientated fibres (cards, webs, slivers, rovings)U.K.
From randomly selected parts of the laboratory bulk sample cut not less than 10 cross-sections each of mass approximately 1 g. Subject the laboratory test sample so formed to the pre-treatment. Recombine the cross-sections by laying them side by side and obtain the test specimen by cutting through them so as to take a portion of each of the 10 lengths.
5.SAMPLING YARNU.K.
5.1.Yarn in packages or in banksU.K.
Sample all the packages in the bulk laboratory sample.
Withdraw the appropriate continuous equal lengths from each package either by winding skeins of the same number of turns on a wrap-reel(6), or by some other means. Unite the lengths side by side either as a single skein or as a tow to form the laboratory test sample, ensuring that there are equal lengths from each package in the skein or tow.
Subject the laboratory test sample to the pre-treatment.
Take test specimens from the laboratory test sample by cutting a bunch of threads of equal length from the skein or tow, taking care to see that the bunch contains all the threads in the sample.
If the tex of the yarn is t and the number of packages selected from the laboratory bulk sample is n, then to obtain a test sample of 10 g, the length of yarn to be withdrawn from each package is 106/nt cm.
If nt is high, i.e. more than 2 000, wind a heavier skein and cut it across in two places to make a tow of suitable mass. The ends of any sample in the form of a tow shall be securely tied before pre-treatment and test specimens taken from a place remote from the tie bands.
5.2.Yarn on warpU.K.
Take the laboratory test sample by cutting a length from the end of the warp, not less than 20 cm long and comprising all the yarns in the warp except the selvedge yarns, which are rejected. Tie the bunch of threads together near one end. If the sample is too large for pre-treatment as a whole divide it into two or more portions, each tied together for pre-treatment, and reunite the portions after each has been pre-treated separately. Take a test specimen by cutting a suitable length from the laboratory test sample from the end remote from the tie band, and comprising all the threads in the warp. For warp of N threads of tex t, the length of a specimen of mass 1 g is 105/Nt cm.
6.SAMPLING FABRICU.K.
6.1.From a laboratory bulk sample consisting of a single cutting representative of the clothU.K.
Cut a diagonal strip from one corner to the other and remove the selvedges. This strip is the laboratory test sample. To obtain a laboratory test sample of x g, the strip area shall be x104/G cm2, where G is the mass of the cloth in g/m2.
Subject the laboratory test sample to the pre-treatment and then cut the strip transversely into four equal lengths and superimpose them. Take test specimens from any part of the layered material by cutting through all the layers so that each specimen contains an equal length of each layer.
If the fabric has a woven design, make the width of the laboratory test sample, measured parallel to the warp direction, not less than one warp repeat of the design. If, with this condition satisfied, the laboratory test sample is too large to be treated as a whole, cut it into equal parts, pre-treat them separately, and superimpose these parts before selection of the test specimen, taking care that corresponding parts of the design do not coincide.
6.2.From a laboratory bulk sample consisting of several cuttingsU.K.
Treat each cutting as described in 6.1, and give each result separately.
7.SAMPLING MADE-UP AND FINISHED PRODUCTSU.K.
The bulk laboratory sample is normally a complete made-up or finished product or representative fraction of one.
Where appropriate determine the percentage of the various parts of the product not having the same fibre content, in order to check compliance with Article 11.
Select a laboratory test sample representative of the part of the made-up or finished product, whose composition must be shown by the label. If the product has several labels, select laboratory test samples representative of each part corresponding to a given label.
If the product whose composition is to be determined is not uniform, it may be necessary to select laboratory test samples from each of the parts of the product and to determine the relative proportions of the various parts in relation to the whole product in question.
Then calculate the percentages taking into account the relative proportions of the sampled parts.
Subject the laboratory test samples to the pre-treatment.
Then select test specimens representative of the pre-treated laboratory test samples.
II. Introduction to the methods for the quantitative analysis of textile fibre mixtures U.K.
Methods for the quantitative analysis of fibre mixtures are based on two main processes, the manual separation and the chemical separation of fibres.
The method of manual separation shall be used whenever possible since it generally gives more accurate results than the chemical method. It can be used for all textiles whose component fibres do not form an intimate mixture, as for example in the case of yarns composed of several elements each of which is made up of only one type of fibre, or fabrics in which the fibre of the warp is of a different kind to that of the weft, or knitted fabrics capable of being unravelled made up of yarns of different types.
In general, the methods of chemical quantitative analysis are based on the selective solution of the individual components. After the removal of a component the insoluble residue is weighed, and the proportion of the soluble component is calculated from the loss in mass. This first part of the Annex gives the information common to the analyses by this method of all fibre mixtures dealt with in the Annex, whatever their composition. It shall thus be used in conjunction with the succeeding individual sections of the Annex, which contain the detailed procedures applicable to particular fibre mixtures. Occasionally, an analysis is based on a principle other than selective solution; in such cases full details are given in the appropriate section.
Mixtures of fibres during processing and, to a lesser extent, finished textiles may contain non-fibrous matter, such as fats, waxes or dressings, or water-soluble matter, either occurring naturally or added to facilitate processing. Non-fibrous matter must be removed before analysis. For this reason a method for removing oils, fats, waxes and water-soluble matter is also given.
In addition, textiles may contain resins or other matter added to confer special properties. Such matter, including dyestuffs in exceptional cases, may interfere with the action of the reagent on the soluble component and/or it may be partially or completely removed by the reagent. This type of added matter may thus cause errors and shall be removed before the sample is analysed. If it is impossible to remove such added matter the methods for quantitative chemical analysis given in this Annex are no longer applicable.
Dye in dyed fabrics is considered to be an integral part of the fibre and is not removed.
Analyses are conducted on the basis of dry mass and a procedure is given for determining dry mass.
The result is obtained by applying to the dry mass of each fibre the agreed allowances listed in Annex IX.
Before proceeding with any analysis, all the fibres present in the mixture shall have been identified. In some methods, the insoluble component of a mixture may be partially dissolved in the reagent used to dissolve the soluble component(s).
Where possible, reagents have been chosen that have little or no effect on the insoluble fibres. If loss in mass is known to occur during the analysis, the result shall be corrected; correction factors for this purpose are given. These factors have been determined in several laboratories by treating, with the appropriate reagent as specified in the method of analysis, fibres cleaned by the pre treatment.
These correction factors apply only to undegraded fibres and different correction factors may be necessary if the fibres have been degraded before or during processing. The procedures given apply to single determinations.
At least two determinations on separate test specimens shall be made, both in the case of manual separation and in the case of chemical separation.
For confirmation, unless technically impossible, it is recommended to use alternative procedures whereby the constituent that was the residue in the standard method is dissolved out first.
CHAPTER 2U.K.
METHODS FOR QUANTITATIVE ANALYSIS OF CERTAIN BINARY TEXTILE FIBRE MIXTURES U.K.
I. General information common to the methods given for the quantitative chemical analysis of textile fibre mixtures U.K.
I.1.FIELD OF APPLICATIONU.K.
The field of application for each method specifies to which fibres the method is applicable.
I.2.PRINCIPLEU.K.
After the identification of the components of a mixture, the non-fibrous material is removed by suitable pre-treatment and then one of the components, usually by selective solution(7). The insoluble residue is weighed and the proportion of soluble component calculated from the loss in mass. Except where this presents technical difficulties, it is preferable to dissolve the fibre present in the greater proportion, thus obtaining the fibre present in the smaller proportion as residue.
I.3.MATERIALS AND EQUIPMENTU.K.
I.3.1.ApparatusU.K.
I.3.1.1.Filter crucibles and weighing bottles large enough to contain such crucibles, or any other apparatus giving identical results.U.K.
I.3.1.2.Vacuum flask.U.K.
I.3.1.3.Desiccator containing self-indicating silica gel.U.K.
I.3.1.4.Ventilated oven for drying specimens at 105 ± 3 °C.U.K.
I.3.1.5.Analytical balance, accurate to 0,0002 g.U.K.
I.3.1.6.Soxhlet extractor or other apparatus giving identical results.U.K.
I.3.2.Reagents.U.K.
I.3.2.1.Light petroleum, redistilled, boiling range 40 to 60 °C.U.K.
I.3.2.2.Other reagents are specified in the appropriate section of each method.U.K.
I.3.2.3.Distilled or deionised water.U.K.
I.3.2.4.Acetone.U.K.
I.3.2.5.Orthophosphoric acid.U.K.
I.3.2.6.Urea.U.K.
I.3.2.7.Sodium bicarbonate.U.K.
All reagents used shall be chemically pure.
I.4.CONDITIONING AND TESTING ATMOSPHEREU.K.
Because dry masses are determined, it is unnecessary to condition the specimen or to conduct analyses in a conditioned atmosphere.
I.5.LABORATORY TEST SAMPLEU.K.
Take a laboratory test sample that is representative of the laboratory bulk sample and sufficient to provide all the specimens, each of at least 1 g, that are required.
I.6.PRE-TREATMENT OF LABORATORY TEST SAMPLE(8) U.K.
Where a substance not to be taken into account in the percentage calculations (see Article 19) is present, it shall first be removed by a suitable method that does not affect any of the fibre constituents.
For this purpose, non-fibrous matter which can be extracted with light petroleum and water is removed by treating the laboratory test sample in a Soxhlet extractor with light petroleum for 1 hour at a minimum rate of six cycles per hour. Allow the light petroleum to evaporate from the sample, which is then extracted by direct treatment consisting in soaking the laboratory test sample in water at room temperature for 1 hour and then soaking it in water at 65 ± 5 °C for a further hour, agitating the liquor from time to time. Use a liquor-laboratory test sample ratio of 100:1. Remove the excess water from the sample by squeezing, suction or centrifuging and then allow the sample to become air-dry.
In the case of elastolefin or fibre mixtures containing elastolefin and other fibres (wool, animal hair, silk, cotton, flax (or linen) true hemp, jute, abaca, alfa, coir, broom, ramie, sisal, cupro, modal, protein, viscose, acrylic, polyamide or nylon, polyester, elastomultiester) the procedure just described shall be slightly modified, in that light petroleum ether shall be replaced by acetone.
In the case of binary fibre mixtures containing elastolefin and acetate the following procedure shall apply as pre-treatment. Extract the laboratory test sample for 10 minutes at 80 °C with a solution containing 25 g/l of 50 % orthophosphoric acid and 50 g/l of urea. Use a liquor-laboratory test sample ratio of 100:1. Wash laboratory test sample in water, then drain and wash it in a 0,1 % sodium bicarbonate solution, finally wash it carefully in water.
Where non-fibrous matter cannot be extracted with light petroleum and water, it shall be removed by substituting for the water method described above a suitable method that does not substantially alter any of the fibre constituents. However, for some unbleached, natural vegetable fibres (e.g. jute, coir) it is to be noted that normal pre-treatment with light petroleum and water does not remove all the natural non-fibrous substances; nevertheless additional pre-treatment is not applied unless the sample contains finishes insoluble in both light petroleum and water.
Analysis reports shall include full details of the methods of pre-treatment used.
I.7.TEST PROCEDUREU.K.
I.7.1.General instructionsU.K.
I.7.1.1.DryingU.K.
Conduct all drying operations for not less than 4 hours and not more than 16 hours at 105 ± 3 °C in a ventilated oven with the oven door closed throughout. If the drying period is less than 14 hours, the specimen must be weighed to check that its mass has become constant. The mass may be considered to have become constant if, after a further drying period of 60 minutes, its variation is less than 0,05 %.
Avoid handling crucibles and weighing bottles, specimens or residues with bare hands during the drying, cooling and weighing operations.
Dry specimens in a weighing bottle with its cover beside it. After drying, stopper the weighing bottle before removing it from the oven, and transfer it quickly to the desiccator.
Dry the filter crucible in a weighing bottle with its cover beside it in the oven. After drying, close the weighing bottle and transfer it quickly to the desiccator.
Where apparatus other than a filter crucible is used, drying operations in the oven shall be conducted in such a way as to enable the dry mass of the fibres to be determined without loss.
I.7.1.2.CoolingU.K.
Conduct all cooling operations in the desiccator, the latter placed beside the balance, until complete cooling of the weighing bottles is attained, and in any case for not less than 2 hours.
I.7.1.3.WeighingU.K.
After cooling, complete the weighing of the weighing bottle within 2 minutes of its removal from the desiccator. Weigh to an accuracy of 0,0002 g.
I.7.2.ProcedureU.K.
Take from the pre-treated laboratory test sample a test specimen weighing at least 1 g. Cut yarn or cloth into lengths of about 10 mm, dissected as much as possible. Dry the specimen in a weighing bottle, cool it in the desiccator and weigh it. Transfer the specimen to the glass vessel specified in the appropriate section of the relevant Union method, reweigh the weighing bottle immediately and obtain the dry mass of the specimen by difference. Complete the test as specified in the appropriate section of the applicable method. Examine the residue microscopically to check that the treatment has in fact completely removed the soluble fibre.
I.8.CALCULATION AND EXPRESSION OF RESULTSU.K.
Express the mass of the insoluble component as a percentage of the total mass of fibre in the mixture. The percentage of soluble component is obtained by difference. Calculate the results on the basis of clean, dry mass, adjusted by (a) the agreed allowances and (b) the correction factors necessary to take account of loss of matter during pre-treatment and analysis. Calculations shall be made by applying the formula given in I.8.2.
I.8.1.Calculation of percentage of insoluble component on clean, dry mass basis, disregarding loss of fibre mass during pre-treatment:U.K.

where
P1%
is the percentage of clean, dry insoluble component,
m
is the dry mass of the test specimen after pre-treatment,
r
is the dry mass of the residue,
d
is the correction factor for loss in mass of the insoluble component in the reagent during the analysis. Suitable values for ‘d’ are given in the relevant section of each method.
Of course, these values for ‘d’ are the normal values applicable to chemically undegraded fibres.
I.8.2.Calculation of percentage of insoluble component on clean, dry mass basis, with adjustment by conventional factors and, where appropriate, correction factors for loss of mass during pre-treatment:U.K.

where
P1A%
is the percentage of insoluble component adjusted by agreed allowances and for loss in mass during pre-treatment,
P1
is the percentage of clean dry insoluble component as calculated from the formula shown in I.8.1,
a1
is the agreed allowance for the insoluble component (see Annex IX),
a2
is the agreed allowance for the soluble component (see Annex IX),
b1
is the percentage loss of insoluble component caused by pre-treatment,
b2
is the percentage loss of soluble component caused by pre-treatment.
The percentage of the second component is P2A % = 100 – P1A %.
Where a special pre-treatment has been used, the values of b1 and b2 shall be determined, if possible, by submitting each of the pure fibre constituents to the pre-treatment applied in the analysis. Pure fibres are those free from all non-fibrous material except that which they normally contain (either naturally or because of the manufacturing process), in the state (unbleached, bleached) in which they are found in the material to be analysed.
Where no clean separate constituent fibres used in the manufacture of the material to be analysed are available, average values of b1 and b2 as obtained from tests performed on clean fibres similar to those in the mixture under examination, shall be used.
If normal pre-treatment by extraction with light petroleum and water is applied, correction factors b1 and b2 may generally be ignored, except in the case of unbleached cotton, unbleached flax (or linen) and unbleached hemp, where the loss due to the pre-treatment is conventionally taken as 4 %, and in the case of polypropylene, where it is taken as 1 %.
In the case of other fibres, losses due to the pre-treatment are conventionally disregarded in calculations.
II. Method of quantitative analysis by manual separation U.K.
II.1.FIELD OF APPLICATIONU.K.
This method is applicable to textile fibres of all types provided they do not form an intimate mixture and that it is possible to separate them by hand.
II.2.PRINCIPLEU.K.
After identification of the constituents of the textile, the non-fibrous material is removed by suitable pre-treatment and then the fibres are separated by hand, dried and weighed in order to calculate the proportion of each fibre in the mixture.
II.3.APPARATUSU.K.
II.3.1.Weighing bottle or any other apparatus giving identical results.U.K.
II.3.2.Desiccator containing self-indicating silica gel.U.K.
II.3.3.Ventilated oven for drying specimens at 105 ± 3 °C.U.K.
II.3.4.Analytical balance, accurate to 0,0002 g.U.K.
II.3.5.Soxhlet extractor, or other apparatus giving an identical result.U.K.
II.3.6.Needle.U.K.
II.3.7.Twist tester or similar apparatus.U.K.
II.4.REAGENTSU.K.
II.4.1.Light petroleum, redistilled, boiling range 40 to 60 °C.U.K.
II.4.2.Distilled or deionised water.U.K.
II.4.3.Acetone.U.K.
II.4.4.Orthophosphoric acid.U.K.
II.4.5.Urea.U.K.
II.4.6.Sodium bicarbonate.U.K.
All reagents used shall be chemically pure.
II.5.CONDITIONING AND TESTING ATMOSPHEREU.K.
See I.4.
II.6.LABORATORY TEST SAMPLEU.K.
See I.5.
II.7.PRE-TREATMENT OF LABORATORY TEST SAMPLEU.K.
See I.6.
II.8.PROCEDUREU.K.
II.8.1.Analysis of yarnU.K.
Select from the pre-treatment laboratory test sample a specimen of mass not less than 1 g. For a very fine yarn, the analysis may be made on a minimum length of 30 m, whatever its mass.
Cut the yarn into pieces of a suitable length and separate the fibre types by means of a needle and, if necessary, a twist tester. The fibre types so obtained are placed in pre-weighed weighing bottles and dried at 105 ± 3 °C until a constant mass is obtained, as described in I.7.1 and I.7.2.
II.8.2.Analysis of clothU.K.
Select from the pre-treated laboratory test sample, well away from all selvedges, a specimen of mass not less than 1 g, with edges carefully trimmed to avoid fraying and running parallel with weft or warp yarns, or in the case of knitted fabrics in the line of wales and courses. Separate the different fibre types, collect them in pre-weighed weighing bottles and proceed as described in II.8.1.
II.9.CALCULATION AND EXPRESSION OF RESULTSU.K.
Express the mass of each fibre constituent as a percentage of the total mass of the fibres in the mixture. Calculate the results on the basis of clean, dry mass, adjusted by (a) the agreed allowances and (b) the correction factors necessary to take account of loss of matter during pre-treatment.
II.9.1.Calculation of percentage masses of clean, dry fibre, disregarding loss of fibre mass during pre-treatment:U.K.

P1%
is the percentage of the first clean, dry component,
m1
is the clean, dry mass of the first component,
m2
is the clean, dry mass of the second component.
II.9.2.For calculation of the percentage of each component with adjustment by agreed allowances and, where appropriate, by correction factors for loss of matter during pre-treatment, see I.8.2.U.K.
III.1.PRECISION OF THE METHODSU.K.
The precision indicated in individual methods relates to the reproducibility.
The reproducibility refers to the reliability, i.e. the closeness of agreement between experimental values obtained by operators in different laboratories or at different times using the same method and obtaining individual results on specimens of an identical consistent mixture.
The reproducibility is expressed by confidence limits of the results for a confidence level of 95 %.
Therefore, the difference between two results in a series of analyses made in different laboratories would, given a normal and correct application of the method to an identical and consistent mixture, exceed the confidence limit only in five cases out of 100.
III.2.TEST REPORTU.K.
III.2.1.State that the analysis was conducted in accordance with this method.U.K.
III.2.2.Give details of any special pre-treatment (see I.6).U.K.
III.2.3.Give the individual results and the arithmetic mean, each to an accuracy of 0,1.U.K.
IV. Special methods U.K.
[F5Summary Table
| a Detailed list of fibres under each method.] | |||
| Method | Field of application a | Reagent | |
|---|---|---|---|
| Soluble component | Insoluble component | ||
| 1. | Acetate | Certain other fibres | Acetone |
| 2. | Certain protein fibres | Certain other fibres | Hypochlorite |
| 3. | Viscose, cupro or certain types of modal | Certain other fibres | Formic acid and zinc chloride |
| 4. | Polyamide or nylon | Certain other fibres | Formic acid, 80 % m/m |
| 5. | Acetate | Certain other fibres | Benzyl alcohol |
| 6. | Triacetate or polylactide | Certain other fibres | Dichloromethane |
| 7. | Certain cellulose fibres | Certain other fibres | Sulphuric acid, 75 % m/m |
| 8. | Acrylics, certain modacrylics or certain chlorofibres | Certain other fibres | Dimethylformamide |
| 9. | Certain chlorofibres | Certain other fibres | Carbon disulphide/acetone, 55,5/44,5 v/v |
| 10. | Acetate | Certain other fibres | Glacial acetic acid |
| 11. | Silk, polyamide or nylon | Certain other fibres | Sulphuric acid, 75 % m/m |
| 12. | Jute | Certain animal fibres | Nitrogen content method |
| 13. | Polypropylene | Certain other fibres | Xylene |
| 14. | Certain fibres | Certain other fibres | Concentrated sulphuric acid method |
| 15. | Chlorofibres, certain modacrylics, certain elastanes, acetates, triacetates | Certain other fibres | Cyclohexanone |
| 16. | Melamine | Certain other fibres | Hot formic acid 90 % m/m |
| [F217. | Polyester | Certain other fibres | Trichloroacetic acid and chloroform] |
Textual Amendments
F5 Substituted by Commission Delegated Regulation (EU) No 286/2012 of 27 January 2012 amending, in order to include a new textile fibre name, Annex I, and, for the purposes of their adaptation to technical progress, Annexes VIII and IX to Regulation (EU) No 1007/2011 of the European Parliament and of the Council on textile fibre names and related labelling and marking of the fibre composition of textile products (Text with EEA relevance).
METHOD No 1U.K. ACETATE AND CERTAIN OTHER FIBRES U.K.(Acetone method)U.K.
1.FIELD OF APPLICATIONU.K.
This method is applicable, after removal of non-fibrous matter, to binary fibre mixtures of:
1.
acetate (19)
with
2.
[F3wool (1), animal hair (2 and 3), silk (4), cotton (5), flax (7) true hemp (8), jute (9), abaca (10), alfa (11), coir (12), broom (13), ramie (14), sisal (15), cupro (21), modal (22), protein (23), viscose (25), acrylic (26), polyamide or nylon (30), polyester (35), polypropylene (37), elastomultiester (45), elastolefin (46), melamine (47), polypropylene/polyamide bicomponent (49) and polyacrylate (50).
In no circumstances is the method applicable to acetate fibres which have been deacetylated on the surface.]
2.PRINCIPLEU.K.
The acetate is dissolved out from a known dry mass of the mixture, with acetone. The residue is collected, washed, dried and weighed; its mass, corrected if necessary, is expressed as a percentage of the dry mass of the mixture. The percentage of dry acetate is found by difference.
3.APPARATUS AND REAGENTS (additional to those specified in the general instructions)U.K.
3.1.ApparatusU.K.
Glass-stoppered conical flasks of at least 200 ml capacity.
3.2.ReagentU.K.
Acetone.
4.TEST PROCEDUREU.K.
Follow the procedure described in the general instructions and proceed as follows:
To the test specimen contained in a glass-stoppered conical flask of at least 200 ml capacity, add 100 ml of acetone per gram of test specimen, shake the flask, stand it for 30 minutes at room temperature, stirring from time to time, and then decant the liquid through the weighed filter crucible.
Repeat the treatment twice more (making three extractions in all), but for periods of 15 minutes only, so that the total time of treatment in acetone is 1 hour. Transfer the residue to the filter crucible. Wash the residue in the filter crucible with acetone and drain with suction. Refill the crucible with acetone and allow to drain under gravity.
Finally, drain the crucible with suction, dry the crucible and residue, and cool and weigh them.
[F35. CALCULATION AND EXPRESSION OF RESULTS U.K.
Calculate the results as described in the general instructions. The value of ‘ d ’ is 1,00, except for melamine and polyacrylate, for which ‘ d ’ is 1,01.]
6.PRECISIONU.K.
On a homogeneous mixture of textile materials, the confidence limits of results obtained by this method are not greater than ± 1 for a confidence level of 95 %.
METHOD No 2U.K. CERTAIN PROTEIN FIBRES AND CERTAIN OTHER FIBRES U.K.(Method using hypochlorite)U.K.
1.FIELD OF APPLICATIONU.K.
This method is applicable, after removal of non-fibrous matter, to binary fibre mixtures of:
1.
certain protein fibres, namely: wool (1), animal hair (2 and 3), silk (4), protein (23)
with
2.
[F5cotton (5), cupro (21), viscose (25), acrylic (26), chlorofibres (27), polyamide or nylon (30), polyester (35), polypropylene (37), elastane (43), glass fibre (44) elastomultiester (45), elastolefin (46), melamine (47) and polypropylene/polyamide bicomponent (49).
If different protein fibres are present, the method gives the total of their amounts but not their individual quantities.]
2.PRINCIPLEU.K.
The protein fibre is dissolved out from a known dry mass of the mixture, with a hypochlorite solution. The residue is collected, washed, dried and weighed; its mass, corrected if necessary, is expressed as a percentage of the dry mass of the mixture. The percentage of dry protein fibre is found by difference.
Either lithium hypochlorite or sodium hypochlorite can be used for the preparation of the hypochlorite solution.
Lithium hypochlorite is recommended in cases involving a small number of analyses or for analyses conducted at fairly lengthy intervals. This is because the percentage of hypochlorite in solid lithium hypochlorite — unlike that in sodium hypochlorite — is virtually constant. If the percentage of hypochlorite is known, hypochlorite content need not be checked iodometrically for each analysis, since a constant weighed portion of lithium hypochlorite can be employed.
3.APPARATUS AND REAGENTS (other than those specified in the general instructions)U.K.
3.1.ApparatusU.K.
(a)Erlenmeyer flask with ground-glass stopper, 250 ml.U.K.
(b)Thermostat, adjustable to 20 ± 2 °C.U.K.
3.2.ReagentsU.K.
(a)Hypochlorite reagentU.K.
(i)Lithium hypochlorite solutionU.K.
This consists of a freshly prepared solution containing 35 ± 2 g/l of active chlorine (approximately 1 M), to which 5 ± 0,5 g/l of previously dissolved sodium hydroxide is added. To prepare, dissolve 100 grams of lithium hypochlorite containing 35 % active chlorine (or 115 grams containing 30 % active chlorine) in approximately 700 ml of distilled water, add 5 grams of sodium hydroxide dissolved in approximately 200 ml of distilled water and make up to 1 litre with distilled water. The solution which has been freshly prepared need not be checked iodometrically.
(ii)Sodium hypochlorite solutionU.K.
This consists of a freshly prepared solution containing 35 ± 2 g/l of active chlorine (approximately 1 M) to which 5 ± 0,5 g/l of previously dissolved sodium hydroxide is added.
Check the active chlorine content of the solution iodometrically before each analysis.
(b)Acetic acid, dilute solutionU.K.
Dilute 5 ml of glacial acetic acid to 1 litre with water.
4.TEST PROCEDUREU.K.
Follow the procedure described in the general instructions and proceed as follows: mix approximately 1 gram of the test specimen with approximately 100 ml of the hypochlorite solution (lithium or sodium hypochlorite) in the 250 ml flask and agitate thoroughly in order to wet out the test specimen.
Then heat the flask for 40 minutes in a thermostat at 20 °C and agitate continuously, or at least at regular intervals. Since the dissolution of the wool proceeds exothermically, the reaction heat of this method must be distributed and removed. Otherwise, considerable errors may be caused by the incipient dissolution of the non-soluble fibres.
After 40 minutes, filter the flask contents through a weighed glass-filter crucible and transfer any residual fibres into the filter crucible by rinsing the flask with a little hypochlorite reagent. Drain the crucible with suction and wash the residue successively with water, dilute acetic acid, and finally water, draining the crucible with suction after each addition. Do not apply suction until each washing liquor has drained under gravity.
Finally, drain the crucible with suction, dry the crucible with the residue, and cool and weigh them.
5.CALCULATION AND EXPRESSION OF RESULTSU.K.
Calculate the results as described in the general instructions. The value of ‘d’ is 1,00, except for cotton, viscose, modal and melamine for which ‘d’ = 1,01, and unbleached cotton, for which ‘d’ = 1,03.
6.PRECISIONU.K.
On homogeneous mixtures of textile materials, the confidence limits for results obtained by this method are not greater than ± 1 for a confidence level of 95 %.
METHOD No 3U.K. [F5VISCOSE, CUPRO OR CERTAIN TYPES OF MODAL AND CERTAIN OTHER FIBRES U.K. (Method using formic acid and zinc chloride)] U.K.
1.FIELD OF APPLICATIONU.K.
This method is applicable, after removal of non-fibrous matter, to binary fibre mixtures of:
1.
viscose (25) or cupro (21), including certain types of modal fibre (22)
with
2.
[F5cotton (5), polypropylene (37), elastolefin (46) and melamine (47).
If a modal fibre is found to be present, a preliminary test shall be carried out to see whether it is soluble in the reagent.
This method is not applicable to mixtures in which the cotton has suffered extensive chemical degradation nor when the viscose or cupro is rendered incompletely soluble by the presence of certain dyes or finishes that cannot be removed completely.]
2.PRINCIPLEU.K.
The viscose, cupro or modal fibre is dissolved from a known dry mass of the mixture, with a reagent consisting of formic acid and zinc chloride. The residue is collected, washed, dried and weighed; its corrected mass is expressed as a percentage of the dry mass of the mixture. The percentage of dry viscose, cupro or modal fibre is found by difference.
3.APPARATUS AND REAGENTS (other than those specified in the general instructions)U.K.
3.1.ApparatusU.K.
(a)Glass-stoppered conical flasks of at least 200 ml capacity.U.K.
(b)Apparatus for maintaining flasks at 40 ± 2 °C.U.K.
3.2.ReagentsU.K.
(a)Solution containing 20 g of fused anhydrous zinc chloride and 68 g of anhydrous formic acid made up to 100 g with water (namely 20 parts by mass of fused anhydrous zinc chloride to 80 parts by mass of 85 % m/m formic acid).U.K.
Note:U.K.
Attention is drawn, in this respect, to point I.3.2.2, which lays down that all reagents used shall be chemically pure; in addition, it is essential to use only fused anhydrous zinc chloride.
(b)Ammonium hydroxide solution: dilute 20 ml of a concentrated ammonia solution (relative density at 20 °C: 0,880) to 1 litre with water.U.K.
4.TEST PROCEDUREU.K.
Follow the procedure described in the general instructions and proceed as follows: place the specimen immediately in the flask, pre-heated to 40 °C. Add 100 ml of the solution of formic acid and zinc chloride, pre-heated to 40 °C per gram of specimen. Insert the stopper and shake the flask vigorously. Keep the flask and its contents at a constant temperature of 40 °C for 2,5 hours, shaking the flask at hourly intervals.
Filter the contents of the flask through the weighed filter crucible and with the help of the reagent transfer to the crucible any fibres remaining in the flask. Rinse with 20 ml of reagent pre-heated to 40 °C.
Wash crucible and residue thoroughly with water at 40 °C. Rinse the fibrous residue in approximately 100 ml of cold ammonia solution (3.2(b)) ensuring that this residue remains wholly immersed in the solution for 10 minutes(9); then rinse thoroughly with cold water.
Do not apply suction until each washing liquor has drained under gravity.
Finally, drain the remaining liquid with suction, dry the crucible and residue, and cool and weigh them.
[F55. CALCULATION AND EXPRESSION OF RESULTS U.K.
Calculate the results as described in the general instructions. The value of ‘ d ’ is 1,00, except for cotton, for which ‘ d ’ = 1,02 and for melamine, for which ‘ d ’ = 1,01.]
6.PRECISIONU.K.
On a homogeneous mixture of textile materials, the confidence limits of results obtained by this method are not greater than ± 2 for a confidence level of 95 %.
METHOD No 4U.K. POLYAMIDE OR NYLON, AND CERTAIN OTHER FIBRES U.K.(Method using 80 % m/m formic acid)U.K.
1.FIELD OF APPLICATIONU.K.
This method is applicable, after removal of non-fibrous matter, to binary fibre mixtures of:
1.
polyamide or nylon (30)
with
2.
wool (1), animal hair (2 and 3), cotton (5), cupro (21), modal (22), viscose (25), acrylic (26), chlorofibre (27), polyester (35), polypropylene (37), glass fibre (44), elastomultiester (45), elastolefin (46) and melamine (47).
As mentioned above, this method is also applicable to mixtures with wool, but when the wool content exceeds 25 %, method No 2 shall be applied (dissolving wool in a solution of alkaline sodium hypochlorite or lithium hypochlorite).
2.PRINCIPLEU.K.
The polyamide or nylon fibre is dissolved out from a known dry mass of the mixture, with formic acid. The residue is collected, washed, dried and weighed; its mass, corrected if necessary, is expressed as a percentage of the dry mass of the mixture. The percentage of dry polyamide or nylon is found by difference.
3.APPARATUS AND REAGENTS (other than those specified in the general instructions)U.K.
3.1.ApparatusU.K.
Glass-stoppered conical flask of at least 200 ml capacity.
3.2.ReagentsU.K.
(a)Formic acid (80 % m/m, relative density at 20 °C: 1,186). Dilute 880 ml of 90 % m/m formic acid (relative density at 20 °C: 1,204) to 1 litre with water. Alternatively, dilute 780 ml of 98 to 100 % m/m formic acid (relative density at 20 °C: 1,220) to 1 litre with water.U.K.
The concentration is not critical within the range 77 to 83 % m/m formic acid.
(b)Ammonia, dilute solution: dilute 80 ml of concentrated ammonia solution (relative density at 20 °C: 0,880) to 1 litre with water.U.K.
4.TEST PROCEDUREU.K.
Follow the procedure described in the general instructions and proceed as follows: to the specimen contained in the conical flask of at least 200 ml capacity, add 100 ml of formic acid per gram of specimen. Insert the stopper, shake the flask to wet out the specimen. Stand the flask for 15 minutes at room temperature, shaking it at intervals. Filter the contents of the flask through the weighed filter crucible and transfer any residual fibres to the crucible by washing out the flask with a little formic acid reagent.
Drain the crucible with suction and wash the residue on the filter successively with formic acid reagent, hot water, dilute ammonia solution, and finally cold water, draining the crucible with suction after each addition. Do not apply suction until each washing liquor has drained under gravity.
Finally, drain the crucible with suction, dry the crucible and residue, and cool and weigh them.
5.CALCULATION AND EXPRESSION OF RESULTSU.K.
Calculate the results as described in the general instructions. The value of ‘d’ is 1,00, except for melamine, for which ‘d’ = 1,01.
6.PRECISIONU.K.
On a homogeneous mixture of textile materials, the confidence limits of results obtained by this method are not greater than ± 1 for a confidence level of 95 %.
METHOD No 5U.K. [F5ACETATE AND CERTAIN OTHER FIBRES U.K. (Method using benzyl alcohol)] U.K.
1.FIELD OF APPLICATIONU.K.
This method is applicable, after removal of non-fibrous matter, to binary fibre mixtures of:
1.
acetate (19)
with
2.
[F3triacetate (24), polypropylene (37), elastolefin (46), melamine (47), polypropylene/polyamide bicomponent (49) and polyacrylate (50).]
2.PRINCIPLEU.K.
The acetate fibre is dissolved out from a known dry mass of the mixture, with benzyl alcohol at 52 ± 2 °C.
The residue is collected, washed, dried and weighed; its mass is expressed as a percentage of the dry mass of the mixture. The percentage of dry acetate is found by difference.
3.APPARATUS AND REAGENTS (other than those specified in the general instructions)U.K.
3.1.ApparatusU.K.
(a)Glass-stoppered conical flask of at least 200 ml capacity.U.K.
(b)Mechanical shaker.U.K.
(c)Thermostat or other apparatus for keeping the flask at a temperature of 52 ± 2 °C.U.K.
3.2.ReagentsU.K.
(a)Benzyl alcohol.U.K.
(b)Ethanol.U.K.
4.TEST PROCEDUREU.K.
Follow the procedure described in the general instructions and proceed as follows:
To the specimen contained in the conical flask, add 100 ml of benzyl alcohol per gram of specimen. Insert the stopper, secure the flask to the shaker so that it is immersed in the water-bath, kept at 52 ± 2 °C, and shake for 20 minutes at this temperature.
(Instead of using a mechanical shaker, the flask may be shaken vigorously by hand).
Decant the liquid through the weighed filter crucible. Add a further dose of benzyl alcohol in the flask and shake as before at 52 ± 2 °C for 20 minutes.
Decant the liquid through the crucible. Repeat the cycle of operations a third time.
Finally pour the liquid and the residue into the crucible; wash any remaining fibres from the flask into the crucible with an extra quantity of benzyl alcohol at 52 ± 2 °C. Drain the crucible thoroughly.
Transfer the fibres into a flask, rinse with ethanol and after shaking manually decant through the filter crucible.
Repeat this rinsing operation two or three times. Transfer the residue into the crucible and drain thoroughly. Dry the crucible and the residue and cool and weigh them.
5.CALCULATION AND EXPRESSION OF RESULTSU.K.
Calculate the results as described in the general instructions. The value of ‘d’ is 1,00, except for melamine, for which ‘d’ = 1,01.
6.PRECISIONU.K.
On a homogeneous mixture of textile materials, the confidence limits of results obtained by this method are not greater than ± 1 for a confidence level of 95 %.
METHOD No 6U.K. [F5TRIACETATES OR POLYLACTIDE AND CERTAIN OTHER FIBRES U.K. (Method using dichloromethane)] U.K.
1.FIELD OF APPLICATIONU.K.
This method is applicable, after removal of non-fibrous matter, to binary fibre mixtures of:
1.
triacetate (24) or polylactide (34)
with
2.
[F3wool (1), animal hair (2 and 3), silk (4), cotton (5), cupro (21), modal (22), viscose (25), acrylic (26), polyamide or nylon (30), polyester (35), polypropylene (37), glass fibre (44), elastomultiester (45), elastolefin (46), melamine (47), polypropylene/polyamide bicomponent (49) and polyacrylate (50).
Note: U.K.
Triacetate fibres which have received a finish leading to partial hydrolysis cease to be completely soluble in the reagent. In such cases, the method is not applicable.]
2.PRINCIPLEU.K.
The triacetate or polylactide fibres are dissolved out from a known dry mass of the mixture, with dichloromethane. The residue is collected, washed, dried and weighed; its mass, corrected if necessary, is expressed as a percentage of the dry mass of the mixture. The percentage of dry triacetate or polylactide is found by difference.
3.APPARATUS AND REAGENTS (other than those specified in the general instructions)U.K.
3.1.ApparatusU.K.
Glass-stoppered conical flask of at least 200 ml capacity.
3.2.ReagentU.K.
Dichloromethane.
4.TEST PROCEDUREU.K.
Follow the procedure described in the general instructions and proceed as follows:
To the test specimen contained in the 200 ml glass-stoppered conical flask, add 100 ml of dichloromethane per gram of the test specimen, insert the stopper, shake the flask to wet out the test specimen and stand for 30 minutes at room temperature, shaking the flask every 10 minutes. Decant the liquid through the weighed filter crucible. Add 60 ml of dichloromethane to the flask containing the residue, shake manually and filter the contents of the flask through the filter crucible. Transfer the residual fibres to the crucible by washing out the flask with a little more dichloromethane. Drain the crucible with suction to remove excess liquid, refill the crucible with dichloromethane and allow it to drain under gravity.
Finally, apply suction to eliminate excess liquid, then treat the residue with boiling water to eliminate all the solvent, apply suction, dry the crucible and residue, cool and weigh them.
5.CALCULATION AND EXPRESSION OF RESULTSU.K.
Calculate the results as described in the general instructions. The value of ‘d’ is 1,00, except in the case of polyester, elastomultiester, elastolefin and melamine for which the value of ‘d’ is 1,01.
6.PRECISIONU.K.
On a homogeneous mixture of textile materials, the confidence limits of results obtained by this method are not greater than ± 1 for a confidence level of 95 %.
METHOD No 7U.K. [F5CERTAIN CELLULOSE FIBRES AND CERTAIN OTHER FIBRES U.K. (Method using 75 % m/m sulphuric acid)] U.K.
1.FIELD OF APPLICATIONU.K.
This method is applicable, after removal of non-fibrous matter, to binary fibre mixtures of:
1.
cotton (5), flax (or linen) (7), true hemp (8), ramie (14), cupro (21), modal (22), viscose (25)
with
2.
[F5polyester (35), polypropylene (37), elastomultiester (45), elastolefin (46) and polypropylene/polyamide bicomponent (49).]
2.PRINCIPLEU.K.
The cellulose fibre is dissolved out from a known dry mass of the mixture, with 75 % m/m sulphuric acid. The residue is collected, washed, dried and weighed; its mass is expressed as a percentage of the dry mass of the mixture. The proportion of dry cellulose fibre is found by difference.
3.APPARATUS AND REAGENTS (other than those specified in the general instructions)U.K.
3.1.ApparatusU.K.
(a)Glass-stoppered conical flask of at least 500 ml capacity.U.K.
(b)Thermostat or other apparatus for maintaining the flask at 50 ± 5 °C.U.K.
3.2.ReagentsU.K.
(a)Sulphuric acid, 75 ± 2 % m/mU.K.
Prepare by adding carefully, while cooling, 700 ml of sulphuric acid (relative density at 20 °C: 1,84) to 350 ml of distilled water.
After the solution has cooled to room temperature, dilute to 1 litre with water.
(b)Ammonia, dilute solutionU.K.
Dilute 80 ml of ammonia solution (relative density at 20 °C: 0,880) to 1 litre with water.
4.TEST PROCEDUREU.K.
Follow the procedure described in the general instructions and proceed as follows:
To the specimen contained in the glass-stoppered conical flask of at least 500 ml capacity, add 200 ml of 75 % sulphuric acid per gram of specimen, insert the stopper and carefully shake the flask to wet out the specimen.
Maintain the flask at 50 ± 5 °C for 1 hour, shaking it at regular intervals of approximately 10 minutes. Filter the contents of the flask through the weighed filter crucible by means of suction. Transfer any residual fibres by washing out the flask with a little 75 % sulphuric acid. Drain the crucible with suction and wash the residue on the filter once by filling the crucible with a fresh portion of sulphuric acid. Do not apply suction until the acid has drained under gravity.
Wash the residue successively several times with cold water, twice with dilute ammonia solution, and then thoroughly with cold water, draining the crucible with suction after each addition. Do not apply suction until each washing liquor has drained under gravity. Finally, drain the remaining liquid from the crucible with suction, dry the crucible and residue, and cool and weigh them.
[F55. CALCULATION AND EXPRESSION OF RESULTS U.K.
Calculate the results as described in the general instructions. The value of ‘ d ’ is 1,00, except for polypropylene/polyamide bicomponent, for which the value of ‘ d ’ is 1,01.]
6.PRECISIONU.K.
On a homogeneous mixture of textile materials, the confidence limits of results obtained by this method are not greater than ± 1 for a confidence level of 95 %.
METHOD No 8U.K. ACRYLICS, CERTAIN MODACRYLICS OR CERTAIN CHLOROFIBRES AND CERTAIN OTHER FIBRES U.K.(Method using dimethylformamide)U.K.
1.FIELD OF APPLICATIONU.K.
This method is applicable, after removal of non-fibrous matter, to binary fibre mixtures of:
2.
[F3wool (1), animal hair (2 and 3), silk (4), cotton (5), cupro (21), modal (22), viscose (25), polyamide or nylon (30), polyester (35), polypropylene (37), elastomultiester (45), elastolefin (46), melamine (47), polypropylene/polyamide bicomponent (49) and polyacrylate (50).
It is equally applicable to acrylics, and certain modacrylics, treated with pre-metallised dyes, but not to those dyed with afterchrome dyes.]
2.PRINCIPLEU.K.
The acrylic, modacrylic or chlorofibre is dissolved out from a known dry mass of the mixture, with dimethylformamide heated in a water-bath at boiling point. The residue is collected, washed, dried and weighed. Its mass, corrected if necessary, is expressed as a percentage of the dry mass of the mixture and the percentage of dry acrylic, modacrylic or chlorofibre is found by difference.
3.APPARATUS AND REAGENTS (other than those specified in the general instructions)U.K.
3.1.ApparatusU.K.
(a)Glass-stoppered conical flask of at least 200 ml capacity.U.K.
(b)Water bath at boiling point.U.K.
3.2.ReagentU.K.
Dimethylformamide (boiling point 153 ± 1 °C) not containing more than 0,1 % water.
This reagent is toxic and the use of a hood is thus recommended.
4.TEST PROCEDUREU.K.
Follow the procedure described in the general instructions and proceed as follows:
To the specimen contained in the glass-stoppered conical flask of at least 200 ml capacity, add per gram of specimen 80 ml of dimethylformamide, pre-heated in the water-bath at boiling point, insert the stopper, shake the flask to wet out the specimen and heat in the water-bath at boiling point for 1 hour. Shake the flask and its contents gently by hand five times during this period.
Decant the liquid through the weighed filter crucible, retaining the fibres in the flask. Add a further 60 ml of dimethylformamide to the flask and heat for a further 30 minutes, shaking the flask and contents gently by hand twice during this period.
Filter the contents of the flask through the filter crucible by means of suction.
Transfer any residual fibre to the crucible by washing out the beaker with dimethylformamide. Drain the crucible with suction. Wash the residue with about 1 litre of hot water at 70-80 °C, filling the crucible each time.
After each addition of water, apply suction briefly but not until the water has drained under gravity. If the washing liquor drains through the crucible too slowly slight suction may be applied.
Finally dry the crucible with the residue, cool and weigh them.
[F35. CALCULATION AND EXPRESSION OF RESULTS U.K.
Calculate the results as described in the general instructions. The value of ‘ d ’ is 1,00, except in the case of wool, cotton, cupro, modal, polyester, elastomultiester, melamine and polyacrylate, for which ‘ d ’ is 1,01.]
6.PRECISIONU.K.
On a homogeneous mixture of textile materials, the confidence limits of results obtained by this method are not greater than ± 1 for a confidence level of 95 %.
METHOD No 9U.K. CERTAIN CHLOROFIBRES AND CERTAIN OTHER FIBRES U.K.(Method using 55,5/44,5 % v/v mixture of carbon disulphide and acetone)U.K.
1.FIELD OF APPLICATIONU.K.
This method is applicable, after removal of non-fibrous matter, to binary fibre mixtures of:
1.
certain chlorofibres (27), namely certain polyvinyl chloride fibres, whether after-chlorinated or not(11)
with
2.
[F3wool (1), animal hair (2 and 3), silk (4), cotton (5), cupro (21), modal (22), viscose (25), acrylic (26), polyamide or nylon (30), polyester (35), polypropylene (37), glass fibre (44), elastomultiester (45), melamine (47), polypropylene/polyamide bicomponent (49) and polyacrylate (50).
When the wool or silk content of the mixture exceeds 25 %, method No 2 shall be used.
When the polyamide or nylon content of the mixture exceeds 25 %, method No 4 shall be used.]
2.PRINCIPLEU.K.
The chlorofibre is dissolved out from a known dry mass of the mixture, with an azeotropic mixture of carbon disulphide and acetone. The residue is collected, washed, dried and weighed; its mass, corrected if necessary, is expressed as a percentage of the dry mass of the mixture. The percentage of dry polyvinyl chloride fibre is found by difference.
3.APPARATUS AND REAGENTS (other than those specified in the general instructions)U.K.
3.1.ApparatusU.K.
(a)Glass-stoppered conical flask of at least 200 ml capacity.U.K.
(b)Mechanical shaker.U.K.
3.2.ReagentsU.K.
(a)Azeotropic mixture of carbon disulphide and acetone (55,5 % by volume carbon disulphide to 44,5 % acetone). As this reagent is toxic, the use of a hood is recommended.U.K.
(b)Ethanol (92 % by volume) or methanol.U.K.
4.TEST PROCEDUREU.K.
Follow the procedure described in the general instructions and proceed as follows:
To the specimen contained in the glass-stoppered conical flask of at least 200 ml capacity, add 100 ml of the azeotropic mixture per gram of specimen. Seal the flask securely, and shake the flask on a mechanical shaker, or vigorously by hand, for 20 minutes at room temperature.
Decant the supernatant liquid through the weighed filter crucible.
Repeat the treatment with 100 ml of fresh reagent. Continue this cycle of operations until no polymer deposit is left on a watch glass when a drop of the extraction liquid is evaporated. Transfer the residue to the filter crucible using more reagent, apply suction to remove the liquid, and rinse the crucible and residue with 20 ml of alcohol and then three times with water. Allow the washing liquor to drain under gravity before draining with suction. Dry the crucible and residue and cool and weigh them.
Note:U.K.
With certain mixtures having a high chlorofibre content there may be substantial shrinkage of the specimen during the drying procedure, as a result of which the dissolution of chlorofibre by the solvent is retarded.
This does not, however, affect the ultimate dissolution of the chlorofibre in the solvent.
[F35. CALCULATION AND EXPRESSION OF RESULTS U.K.
Calculate the results as described in the general instructions. The value of ‘ d ’ is 1,00, except for melamine and polyacrylate, for which ‘ d ’ is 1,01.]
6.PRECISIONU.K.
On a homogeneous mixture of textile materials, the confidence limits of the results obtained by this method are not greater than ± 1 for a confidence level of 95 %.
METHOD No 10U.K. [F5ACETATE AND CERTAIN OTHER FIBRES U.K. (Method using glacial acetic acid)] U.K.
1.FIELD OF APPLICATIONU.K.
This method is applicable, after removal of non-fibrous matter, to binary fibre mixtures of:
1.
acetate (19)
with
2.
[F5certain chlorofibres (27) namely polyvinyl chloride fibres, whether after-chlorinated or not, polypropylene (37), elastolefin (46), melamine (47) and polypropylene/polyamide bicomponent (49).]
2.PRINCIPLEU.K.
The acetate fibre is dissolved out from a known dry mass of the mixture, with glacial acetic acid. The residue is collected, washed, dried and weighed; its mass, corrected if necessary, is expressed as a percentage of the dry mass of the mixture. The percentage of dry acetate is found by difference.
3.APPARATUS AND REAGENTS (other than those specified in the general instructions)U.K.
3.1.ApparatusU.K.
(a)Glass-stoppered conical flask of at least 200 ml capacity.U.K.
(b)Mechanical shaker.U.K.
3.2.ReagentU.K.
Glacial acetic acid (over 99 %). This reagent shall be handled with care since it is highly caustic.
4.TEST PROCEDUREU.K.
Follow the procedure described in the general instructions and proceed as follows:
To the specimen contained in the glass-stoppered conical flask of at least 200 ml capacity, add 100 ml glacial acetic acid per gram of specimen. Seal the flask securely and shake on the mechanical shaker, or vigorously by hand, for 20 minutes at room temperature. Decant the supernatant liquid through the weighed filter crucible. Repeat this treatment twice, using 100 ml of fresh reagent each time, making three extractions in all.
Transfer the residue to the filter crucible, drain with suction to remove the liquid and rinse the crucible and the residue with 50 ml of glacial acetic acid, and then three times with water. After each rinse, allow the liquid to drain under gravity before applying suction. Dry the crucible and residue, and cool and weigh them.
5.CALCULATION AND EXPRESSION OF RESULTSU.K.
Calculate the results as described in the general instructions. The value of ‘d’ is 1,00.
6.PRECISIONU.K.
On a homogeneous mixture of textile materials, the confidence limits of the results obtained by this method are not greater than ± 1 for a confidence level of 95 %.
METHOD No 11U.K. [F5SILK OR POLYAMIDE AND CERTAIN OTHER FIBRES U.K. (Method using 75 % m/m sulphuric acid)] U.K.
[F51. FIELD OF APPLICATION U.K.
This method is applicable, after removal of non-fibrous matter, to binary mixtures of:
1.
silk (4) or polyamide or nylon (30)
with
2.
wool (1), animal hair (2 and 3), polypropylene (37), elastolefin (46), melamine (47) and polypropylene/polyamide bicomponent (49).]
[F52. PRINCIPLE U.K.
The silk or polyamide or nylon fibre is dissolved out from a known dry mass of the mixture, with 75 % m/m sulphuric acid (12) .
The residue is collected, washed, dried and weighed. Its mass, corrected if necessary, is expressed as a percentage of the dry mass of the mixture. The percentage of the dry silk or polyamide or nylon is found by difference.]
3.APPARATUS AND REAGENTS (other than those specified in the general instructions)U.K.
3.1.ApparatusU.K.
Glass-stoppered conical flask of at least 200 ml capacity.
3.2.ReagentsU.K.
(a)Sulphuric acid (75 ± 2 % m/m)U.K.
Prepare by adding carefully, while cooling, 700 ml sulphuric acid (relative density at 20 °C: 1,84) to 350 ml distilled water.
After cooling to room temperature, dilute the solution to 1 litre with water.
(b)Sulphuric acid, dilute solution: add 100 ml sulphuric acid (relative density at 20 °C: 1,84) slowly to 1 900 ml distilled water.U.K.
(c)Ammonia, dilute solution: dilute 200 ml concentrated ammonia (relative density at 20 °C: 0,880) to 1 litre with water.U.K.
[F54. TEST PROCEDURE U.K.
Follow the procedure described in the general instructions and proceed as follows:
To the specimen contained in a glass-stoppered conical flask of at least 200 ml capacity, add 100 ml of 75 % m/m sulphuric acid per gram of specimen and insert the stopper. Shake vigorously and stand for 30 minutes at room temperature. Shake again and stand for 30 minutes. Shake a last time and filter the contents of the flask through the weighed filter crucible. Wash any remaining fibres from the flask with the 75 % sulphuric acid reagent. Wash the residue on the crucible successively with 50 ml of the dilute sulphuric acid reagent, 50 ml water and 50 ml of the dilute ammonia solution. Each time allow the fibres to remain in contact with the liquid for about 10 minutes before applying suction. Finally rinse with water, leaving the fibres in contact with the water for about 30 minutes. Drain the crucible with suction, dry the crucible and residue, and cool and weigh them.
In the case of binary mixtures of polyamide with polypropylene/polyamide bicomponent, after filtering fibres through the weighed filter crucible and before applying the described washing procedure, wash twice the residue on the filter crucible with 50 ml of 75 % sulphuric acid reagent each time.]
[F55. CALCULATION AND EXPRESSION OF RESULTS U.K.
Calculate the results as described in the general instructions. The value of ‘ d ’ is 1,00, except for wool, for which ‘ d ’ = 0,985, for polypropylene/polyamide bicomponent, for which ‘ d ’ = 1,005 and for melamine, for which ‘ d ’ = 1,01.
6. PRECISION U.K.
On a homogeneous mixture of textile materials, the confidence limits of results obtained by this method are not greater than ± 1 for a confidence level of 95 %, except for binary mixtures of polyamide with polypropylene/polyamide bicomponent for which the confidence limits of results are not greater than ± 2.]
METHOD No 12U.K. JUTE AND CERTAIN ANIMAL FIBRES U.K.(Method by determining nitrogen content)U.K.
1.FIELD OF APPLICATIONU.K.
This method is applicable, after removal of non-fibrous matter, to binary fibre mixtures of:
1.
jute (9)
with
2.
certain animal fibres.
The animal-fibre component may consist solely of hair (2 and 3) or wool (1) or of any mixture of the two. This method is not applicable to textile mixtures containing non-fibrous matter (dyes, finishes, etc.) with a nitrogen base.
2.PRINCIPLEU.K.
The nitrogen content of the mixture is determined, and from this and the known or assumed nitrogen contents of the two components, the proportion of each component is calculated.
3.APPARATUS AND REAGENTS (other than those specified in the general instructions)U.K.
3.1.ApparatusU.K.
(a)Kjeldahl digestion flask, 200-300 ml capacity.U.K.
(b)Kjeldahl distillation apparatus with steam injection.U.K.
(c)Titration apparatus, allowing precision of 0,05 ml.U.K.
3.2.ReagentsU.K.
(a)Toluene.U.K.
(b)Methanol.U.K.
(c)Sulphuric acid, relative density at 20 °C: 1,84(13).U.K.
(d)Potassium sulphate(13).U.K.
(e)Selenium dioxide(13).U.K.
(f)Sodium hydroxide solution (400 g/litre). Dissolve 400 g of sodium hydroxide in 400-500 ml of water and dilute to 1 litre with water.U.K.
(g)Mixed indicator. Dissolve 0,1 g of methyl red in 95 ml of ethanol and 5 ml of water, and mix with 0,5 g of bromocresol green dissolved in 475 ml of ethanol and 25 ml of water.U.K.
(h)Boric acid solution. Dissolve 20 g of boric acid in 1 litre of water.U.K.
(i)Sulphuric acid, 0,02N (standard volumetric solution).U.K.
4.PRE-TREATMENT OF TEST SAMPLEU.K.
The following pre-treatment is substituted for the pre-treatment described in the general instructions:
Extract the air-dry laboratory test sample in a Soxhlet apparatus with a mixture of 1 volume of toluene and 3 volumes of methanol for 4 hours at a minimum rate of 5 cycles per hour. Allow the solvent to evaporate from the sample in air, and remove the last traces in an oven at 105 ± 3 °C. Then extract the sample in water (50 ml per g of sample) by boiling under reflux for 30 minutes. Filter, return the sample to the flask, and repeat the extraction with an identical volume of water. Filter, remove excess water from the sample by squeezing, suction, or centrifuging and then allow the sample to become air-dry.
Note:U.K.
The toxic effects of toluene and methanol shall be borne in mind and full precautions shall be taken in their use.
5.TEST PROCEDUREU.K.
5.1.General instructionsU.K.
Follow the procedure described in the general instructions as regards the selection, drying and weighing of the specimen.
5.2.Detailed procedureU.K.
Transfer the specimen to a Kjeldahl digestion flask. To the specimen weighing at least 1 g contained in the digestion flask, add, in the following order, 2,5 g potassium sulphate, 0,1-0,2 g selenium dioxide and 10 ml sulphuric acid (relative density at 20 °C: 1,84). Heat the flask, gently at first, until the whole of the fibre is destroyed, and then heat it more vigorously until the solution becomes clear and almost colourless. Heat it for a further 15 minutes. Allow the flask to cool, dilute the contents carefully with 10-20 ml water, cool, transfer the contents quantitatively to a 200 ml graduated flask and make up to volume with water to form the digest solution. Place about 20 ml of boric acid solution in a 100 ml conical flask and place the flask under the condenser of the Kjeldahl distillation apparatus so that the delivery tube dips just below the surface of the boric acid solution. Transfer exactly 10 ml of digest solution to the distillation flask, add not less than 5 ml of sodium hydroxide solution to the funnel, lift the stopper slightly and allow the sodium hydroxide solution to run slowly into the flask. If the digest solution and sodium hydroxide solution remain as two separate layers, mix them by gentle agitation. Heat the distillation flask gently and pass it into steam from the generator. Collect about 20 ml of distillate, lower the conical flask so that the tip of the delivery tube of the condenser is about 20 mm above the surface of the liquid and distil for 1 minute more. Rinse the tip of the delivery tube with water, catching the washings in the conical flask. Remove the conical flask and replace it with another conical flask containing roughly 10 ml of boric acid solution and collect about 10 ml distillate.
Titrate the two distillates separately with 0,02 N sulphuric acid, use the mixed indicator. Record the total titre for the two distillates. If the titre for the second distillate is more than 0,2 ml, repeat the test and start the distillation again using a fresh aliquot of digest solution.
Carry out a blank determination, i.e. digestion and distillation using the reagents only.
6.CALCULATION AND EXPRESSION OF RESULTSU.K.
6.1.Calculate the percentage nitrogen content in the dry specimen as follows:U.K.
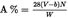
where
A
=
percentage nitrogen in the clean dry specimen,
V
=
total volume in ml of standard sulphuric acid used in the determination,
b
=
total volume in ml of standard sulphuric acid used in the blank determination,
N
=
normality of standard sulphuric acid,
W
=
dry mass (g) of specimen.
6.2.Using the values of 0,22 % for the nitrogen content of jute and 16,2 % for the nitrogen content of animal fibre, both percentages being expressed on the dry mass of the fibre, calculate the composition of the mixture as follows:U.K.

where
PA%
=
percentage of animal fibre in the clean dry specimen.
7.PRECISIONU.K.
On a homogeneous mixture of textile materials, the confidence limits of results obtained by this method are not greater than ± 1 for a confidence level of 95 %.
METHOD No 13U.K. POLYPROPYLENE FIBRES AND CERTAIN OTHER FIBRES U.K.(Xylene method)U.K.
1.FIELD OF APPLICATIONU.K.
This method is applicable, after removal of non-fibrous matter, to binary fibre mixtures of:
1.
polypropylene fibres (37)
with
2.
[F3wool (1), animal hair (2 and 3), silk (4), cotton (5), acetate (19), cupro (21), modal (22), triacetate (24), viscose (25), acrylic (26), polyamide or nylon (30), polyester (35), glass fibre (44), elastomultiester (45), melamine (47) and polyacrylate (50).]
2.PRINCIPLEU.K.
The polypropylene fibre is dissolved out from a known dry mass of the mixture with boiling xylene. The residue is collected, washed, dried and weighed; its mass, corrected if necessary, is expressed as a percentage of the dry mass of the mixture. The percentage of polypropylene is found by difference.
3.APPARATUS AND REAGENTS (other than those specified in the general instructions)U.K.
3.1.ApparatusU.K.
(a)Glass-stoppered conical flask of at least 200 ml capacity.U.K.
(b)Reflux condenser (suitable for liquids of high boiling point), fitting the conical flask (a).U.K.
(c)Heating mantle at boiling point of xylene.U.K.
3.2.ReagentU.K.
Xylene distilling between 137 and 142 °C.
Note:U.K.
Xylene is highly flammable and has a toxic vapour. Suitable precautions must be taken in its use.
4.TEST PROCEDUREU.K.
Follow the procedure described in the general instructions then proceed as follows:
To the specimen contained in the conical flask (3.1(a)), add 100 ml of xylene (3.2) per gram of specimen. Attach the condenser (3.1(b)), bring the contents to the boil and maintain at boiling point for 3 minutes.
Immediately decant the hot liquid through the weighed filter crucible (see Note 1). Repeat this treatment twice more, each time using a fresh 50 ml portion of solvent.
Wash the residue remaining in the flask successively with 30 ml of boiling xylene (twice), then with 75 ml of light petroleum (I.3.2.1 of general instructions) (twice). After the second wash with light petroleum, filter the contents of the flask through the crucible, transfer any residual fibres to the crucible with the aid of a small quantity of light petroleum and allow the solvent to evaporate. Dry the crucible and residue, cool and weigh them.
Notes:U.K.
1.The filter crucible through which the xylene is to be decanted must be pre-heated.U.K.
2.After the treatment with boiling xylene, ensure that the flask containing the residue is cooled sufficiently before the light petroleum is introduced.U.K.
3.In order to reduce the fire and toxicity hazards to the operator, a hot extraction apparatus using the appropriate procedures, giving identical results, may be used(14).U.K.
[F35. CALCULATION AND EXPRESSION OF RESULTS U.K.
Calculate the results as described in the general instructions. The value of ‘ d ’ is 1,00, except for melamine and polyacrylate, for which ‘ d ’ is 1,01.]
6.PRECISIONU.K.
On a homogeneous mixture of textile materials, the confidence limits of results obtained by this method are not greater than ± 1 for a confidence level of 95 %.
METHOD No 14U.K. [F5CERTAIN FIBRES AND CERTAIN OTHER FIBRES U.K. (Method using concentrated sulphuric acid)] U.K.
1.FIELD OF APPLICATIONU.K.
This method is applicable, after removal of non-fibrous matter, to binary fibre mixtures of:
1.
cotton (5), acetate (19), cupro (21), modal (22), triacetate (24), viscose (25), certain acrylics (26), certain modacrylics (29), polyamide or nylon (30), polyester (35) and elastomultiester (45)
with
2.
[F5chlorofibres (27) based on homopolymers of vinyl chloride, whether after-chlorinated or not, polypropylene (37), elastolefin (46), melamine (47) and polypropylene/polyamide bicomponent (49).
The modacrylics concerned are those which give a limpid solution when immersed in concentrated sulphuric acid (relative density 1,84 at 20 °C).
This method can be used in place of methods No 8 and 9.]
[F52. PRINCIPLE U.K.
The constituent other than the chlorofibre, polypropylene, elastolefin, melamine or polypropylene/polyamide bicomponent (i.e. the fibres mentioned in paragraph 1.1) is dissolved out from a known dry mass of the mixture with concentrated sulphuric acid (relative density 1,84 at 20 °C). The residue, consisting of the chlorofibre, polypropylene, elastolefin, melamine or polypropylene/polyamide bicomponent is collected, washed, dried and weighed; its mass, corrected if necessary, is expressed as a percentage of the dry mass of the mixture. The percentage of the second constituents is obtained by difference.]
3.APPARATUS AND REAGENTS (other than those specified in the general instructions)U.K.
3.1.ApparatusU.K.
(a)Glass-stoppered conical flask of at least 200 ml capacity.U.K.
(b)Glass rod with flattened end.U.K.
3.2.ReagentsU.K.
(a)Sulphuric acid, concentrated (relative density at 20 °C: 1,84).U.K.
(b)Sulphuric acid, approximately 50 % (m/m) aqueous solution.U.K.
Prepare by adding carefully, while cooling, 400 ml of sulphuric acid (relative density at 20 °C: 1,84) to 500 ml of distilled or deionised water. After cooling to room temperature, dilute the solution to one litre with water.
(c)Ammonia, dilute solution.U.K.
Dilute 60 ml of concentrated ammonia solution (relative density at 20 °C: 0,880) to one litre with distilled water.
4.TEST PROCEDUREU.K.
Follow the procedure described in the general instructions, then proceed as follows:
To the test specimen contained in the flask (3.1(a)) add 100 ml of sulphuric acid (3.2(a)) per gram of specimen.
Allow the contents of the flask to remain at room temperature for 10 minutes and during that time stir the test specimen occasionally by means of the glass rod. If a woven or knitted fabric is being treated, wedge it between the wall of the flask and the glass rod and exert a light pressure in order to separate the material dissolved by the sulphuric acid.
Decant the liquid through the weighed filter crucible. Add to the flask a fresh portion of 100 ml of sulphuric acid (3.2(a)) and repeat the same operation. Transfer the contents of the flask to the filter crucible and transfer the fibrous residue there with the aid of the glass rod. If necessary, add a little concentrated sulphuric acid (3.2(a)) to the flask in order to remove any fibres adhering to the wall. Drain the filter crucible with suction; remove the filtrate by emptying or changing the filter-flask, wash the residue in the crucible successively with 50 % sulphuric acid solution (3.2(b)), distilled or deionised water (I.3.2.3 of the general instructions), ammonia solution (3.2(c)) and finally wash thoroughly with distilled or deionised water, draining the crucible with suction after each addition. (Do not apply suction during the washing operation, but only after the liquid has drained off by gravity.) Dry the crucible and residue, cool and weigh them.
[F55. CALCULATION AND EXPRESSION OF RESULTS U.K.
Calculate the results as described in the general instructions. The value of ‘ d ’ is 1,00, except for melamine and polypropylene/polyamide bicomponent, for which the value of ‘ d ’ is 1,01.]
6.PRECISIONU.K.
On a homogeneous mixture of textile materials, the confidence limits of results obtained by this method are not greater than ± 1 for a confidence level of 95 %.
METHOD No 15U.K. CHLOROFIBRES, CERTAIN MODACRYLICS, CERTAIN ELASTANES, ACETATES, TRIACETATES AND CERTAIN OTHER FIBRES U.K.(Method using cyclohexanone)U.K.
1.FIELD OF APPLICATIONU.K.
This method is applicable, after removal of non-fibrous matter, to binary fibre mixtures of:
1.
acetate (19), triacetate (24), chlorofibre (27), certain modacrylics (29), certain elastanes (43)
with
2.
[F3wool (1), animal hair (2 and 3), silk (4), cotton (5), cupro (21), modal (22), viscose (25), acrylic (26), polyamide or nylon (30), glass fibre (44), melamine (47) and polyacrylate (50).
Where modacrylics or elastanes are present, a preliminary test shall first be carried out to determine whether the fibre is completely soluble in the reagent.
Mixtures containing chlorofibres may also be analysed by using method No 9 or 14.]
2.PRINCIPLEU.K.
The acetate and triacetate fibres, chlorofibres, certain modacrylics, and certain elastanes are dissolved out from a known dry mass with cyclohexanone at a temperature close to boiling point. The residue is collected, washed, dried and weighed; its mass, corrected if necessary, is expressed as a percentage of the dry mass of the mixture. The percentage of chlorofibre, modacrylic, elastane, acetate and triacetate is found by difference.
3.APPARATUS AND REAGENTS (other than those specified in the general instructions)U.K.
3.1.ApparatusU.K.
(a)Hot extraction apparatus suitable for use in the test procedure in point 4 (see figure: this is a variant of the apparatus described in Melliand Textilberichte 56 (1975) pp. 643-645).U.K.
(b)Filter crucible to contain the test specimen.U.K.
(c)Porous baffle (porosity grade 1).U.K.
(d)Reflux condenser that can be adapted to the distillation flask.U.K.
(e)Heating device.U.K.
3.2.ReagentsU.K.
(a)Cyclohexanone, boiling point 156 °C.U.K.
(b)Ethyl alcohol, 50 % by volume.U.K.
Note:U.K.
Cyclohexanone is flammable and toxic. Suitable precautions must be taken in its use.
4.TEST PROCEDUREU.K.
Follow the procedure described in the general instructions and then proceed as follows:
Pour into the distillation flask 100 ml of cyclohexanone per gram of material, insert the extraction container in which the filter crucible, containing the specimen and the porous baffle, slightly inclined, have previously been placed. Insert the reflux condenser. Bring to the boil and continue extraction for 60 minutes at a minimum rate of 12 cycles per hour.
After extraction and cooling remove the extraction container, take out the filter crucible and remove the porous baffle. Wash the contents of the filter crucible three or four times with 50 % ethyl alcohol heated to about 60 °C and subsequently with 1 litre of water at 60 °C.
Do not apply suction during or between the washing operations. Allow the liquid to drain under gravity and then apply suction.
Finally, dry the crucible with the residue, cool and weigh them.
[F35. CALCULATION AND EXPRESSION OF RESULTS U.K.
Calculate the results as described in the general instructions. The value of ‘ d ’ is 1,00, except in the case of polyacrylate, for which ‘ d ’ is 1,02, silk and melamine, for which ‘ d ’ is 1,01, and acrylic, for which ‘ d ’ is 0,98.]
6.PRECISIONU.K.
On homogeneous mixtures of textile fibres, the confidence limits of results obtained by this method are not greater than ± 1 for a confidence level of 95 %.

METHOD No 16U.K. [F5MELAMINE AND CERTAIN OTHER FIBRES U.K. (Method using hot formic acid)] U.K.
1.FIELD OF APPLICATIONU.K.
This method is applicable, after removal of non-fibrous matter, to binary fibre mixtures of:
1.
melamine (47)
with
2.
[F5cotton (5), aramid (31) and polypropylene (37).]
2.PRINCIPLEU.K.
The melamine is dissolved out from a known dry mass of the mixture with hot formic acid (90 % m/m).
The residue is collected, washed, dried and weighed; its mass, corrected if necessary, is expressed as a percentage of the dry mass of the mixture. The percentage of the second constituents is obtained by difference.
Note:U.K.
Keep strictly the recommended temperature range because the solubility of melamine is very much dependent on temperature.
3.APPARATUS AND REAGENTS (other than those specified in the general instructions)U.K.
3.1.ApparatusU.K.
(a)Glass-stoppered conical flask of at least 200 ml capacity.U.K.
(b)Shaking water bath or other apparatus to shake and maintain the flask at 90 ± 2 °C.U.K.
3.2.ReagentsU.K.
(a)Formic acid (90 % m/m, relative density at 20 °C: 1,204). Dilute 890 ml of 98 to 100 % m/m formic acid (relative density at 20 °C: 1,220) to 1 litre with water.U.K.
Hot formic acid is very corrosive and must be handled with care.
(b)Ammonia, dilute solution: dilute 80 ml of concentrated ammonia solution (relative density at 20 °C: 0,880) to 1 litre with water.U.K.
4.TEST PROCEDUREU.K.
Follow the procedure described in the general instructions, then proceed as follows:
To the test specimen contained in the glass-stoppered conical flask of at least 200 ml capacity, add 100 ml of formic acid per gram of specimen. Insert the stopper and shake the flask to wet out the specimen. Maintain the flask in a shaking water bath at 90 ± 2 °C for 1 hour, shaking it vigorously. Cool the flask to room temperature. Decant the liquid through the weighed filter crucible. Add 50 ml of formic acid to the flask containing the residue, shake manually and filter the contents of the flask through the filter crucible. Transfer any residual fibres to the crucible by washing out the flask with a little more formic acid reagent. Drain the crucible with suction and wash the residue with formic acid reagent, hot water, dilute ammonia solution, and finally cold water, draining the crucible with suction after each addition. Do not apply suction until each washing liquor has drained under gravity. Finally, drain the crucible with suction, dry the crucible and residue, and cool and weigh them.
5.CALCULATION AND EXPRESSION OF RESULTSU.K.
Calculate the results as described in the general instructions. The value of ‘d’ is 1,02.
6.PRECISIONU.K.
On a homogeneous mixture of textile materials, the confidence limits of results obtained by this method are not greater than ± 2 for a confidence level of 95 %.
[F2METHOD No 17 U.K. POLYESTER AND CERTAIN OTHER FIBRES U.K. (Method using trichloroacetic acid and chloroform) U.K.
1. FIELD OF APPLICATION U.K.
This method is applicable, after removal of non-fibrous matter, to binary fibre mixtures of:
1.
polyester (35)
with
2.
polyacrylate (50)
2. GENERAL INFORMATION U.K.
The principle, apparatus and reagent, test procedure, calculation and expression of results that apply to binary fibre mixtures of polyester with polyacrylate are those described in standard EN ISO 1833-25:2013. The ‘ d ’ value is 1,01.]
CHAPTER 3U.K.
QUANTITATIVE ANALYSIS OF TERNARY TEXTILE FIBRE MIXTURES U.K.
INTRODUCTIONU.K.
In general, the methods of quantitative chemical analysis are based on the selective solution of the individual components. There are four possible variants of this method:
1.
Using two different test specimens, a component (a) is dissolved from the first test specimen, and another component (b) from the second test specimen. The insoluble residues of each specimen are weighed and the percentage of each of the two soluble components is calculated from the respective losses in mass. The percentage of the third component (c) is calculated by difference.
2.
Using two different test specimens, a component (a) is dissolved from the first test specimen and two components (a and b) from the second test specimen. The insoluble residue of the first test specimen is weighed and the percentage of the component (a) is calculated from the loss in mass. The insoluble residue of the second test specimen is weighed; it corresponds to component (c). The percentage of the third component (b) is calculated by difference.
3.
Using two different test specimens, two components (a and b) are dissolved from the first test specimen and two components (b and c) from the second test specimen. The insoluble residues correspond to the two components (c) and (a) respectively. The percentage of the third component (b) is calculated by difference.
4.
Using only one test specimen, after removal of one of the components, the insoluble residue formed by the two other fibres is weighed and the percentage of the soluble component is calculated from the loss in mass. One of the two fibres of the residue is dissolved, the insoluble component is weighed and the percentage of the second soluble component is calculated from the loss in mass.
Where a choice is possible, it is advisable to use one of the first three variants.
Where chemical analysis is used, the expert responsible for the analysis must take care to select methods employing solvents which dissolve only the correct fibre(s), leaving the other fibre(s) intact.
By way of example, a table is given in Section V which contains a certain number of ternary fibre mixtures, together with methods for analysing binary fibre mixtures which can, in principle, be used for analysing these ternary fibre mixtures.
In order to reduce the possibility of error to a minimum, it is recommended that, whenever possible, chemical analysis using at least two of the four abovementioned variants shall be made.
Before proceeding with any analysis, all the fibres present in the mixture must be identified. In some chemical methods, the insoluble component of a mixture may be partially dissolved in the reagent used to dissolve the soluble component(s). Wherever possible, reagents have been chosen that have little or no effect on the insoluble fibres. If a loss in mass is known to occur during the analysis, the result shall be corrected; correction factors are given for this purpose. These factors have been determined in several laboratories by treating, with the appropriate reagent as specified in the method of analysis, fibres cleaned by the pre-treatment. These correction factors apply only to undergraded fibres and different correction factors may be necessary if the fibres have been degraded before or during processing. If the fourth variant, in which a textile fibre is subjected to the successive action of two different solvents, must be used, correction factors must be applied for possible losses in mass undergone by the fibre in the two treatments. At least two determinations shall be made, both in the case of manual separation and in the case of chemical separation.
I. General information on methods for the quantitative chemical analysis of ternary fibre mixtures U.K.
Information common to the methods given for the quantitative chemical analysis of ternary fibre mixtures.
I.1.FIELD OF APPLICATIONU.K.
The field of application of each method for analysing binary fibre mixtures specifies to which fibres the method is applicable (see Chapter 2 relating to methods for quantitative analysis of certain binary textile fibre mixtures).
I.2.PRINCIPLEU.K.
After the identification of the components of a mixture, the non-fibrous material is removed by suitable pre-treatment and then one or more of the four variants of the process of selective solution described in the introduction is applied. Except where this presents technical difficulties, it is preferable to dissolve the major fibre component so as to obtain the minor fibre component as final residue.
I.3.MATERIALS AND EQUIPMENTU.K.
I.3.1.ApparatusU.K.
I.3.1.1.Filter crucibles and weighing bottles large enough to contain such crucibles, or any other apparatus giving identical results.U.K.
I.3.1.2.Vacuum flask.U.K.
I.3.1.3.Desiccator containing self-indicating silica gel.U.K.
I.3.1.4.Ventilated oven for drying specimens at 105 ± 3 °C.U.K.
I.3.1.5.Analytical balance, accurate to 0,0002 g.U.K.
I.3.1.6.Soxhlet extractor or other apparatus giving identical results.U.K.
I.3.2.ReagentsU.K.
I.3.2.1.Light petroleum, redistilled, boiling range 40 to 60 °C.U.K.
I.3.2.2.Other reagents are specified in the appropriate sections of each method.U.K.
I.3.2.3.Distilled or deionised water.U.K.
I.3.2.4.Acetone.U.K.
I.3.2.5.Orthophosphoric acid.U.K.
I.3.2.6.Urea.U.K.
I.3.2.7.Sodium bicarbonate.U.K.
All reagents used shall be chemically pure.
I.4.CONDITIONING AND TESTING ATMOSPHEREU.K.
Because dry masses are determined, it is unnecessary to condition the specimen or to conduct analyses in a conditioned atmosphere.
I.5.LABORATORY TEST SAMPLEU.K.
Take a laboratory test sample that is representative of the laboratory bulk sample and sufficient to provide all the specimens, each of at least 1 g, that are required.
I.6.PRE-TREATMENT OF LABORATORY TEST SAMPLE(15) U.K.
Where a substance not to be taken into account in the percentage calculations (see Article 19) is present, it shall first be removed by a suitable method that does not affect any of the fibre constituents.
For this purpose, non-fibrous matter which can be extracted with light petroleum and water is removed by treating the laboratory test sample in a Soxhlet extractor with light petroleum for 1 hour at a minimum rate of six cycles per hour. Allow the light petroleum to evaporate from the laboratory test sample, which is then extracted by direct treatment consisting in soaking the laboratory test sample in water at room temperature for 1 hour and then soaking it in water at 65 ± 5 °C for a further hour, agitating the liquor from time to time. Use a liquor: laboratory test sample ratio of 100:1. Remove the excess water from the laboratory test sample by squeezing, suction or centrifuging and then allow the laboratory test sample to become air-dry.
In the case of elastolefin or fibre mixtures containing elastolefin and other fibres (wool, animal hair, silk, cotton, flax (or linen), true hemp, jute, abaca, alfa, coir, broom, ramie, sisal, cupro, modal, protein, viscose, acrylic, polyamide or nylon, polyester, elastomultiester) the procedure just described shall be slightly modified, in fact light petroleum ether shall be replaced by acetone.
Where non-fibrous matter cannot be extracted with light petroleum and water, it shall be removed by substituting for the water method described above a suitable method that does not substantially alter any of the fibre constituents. However, for some unbleached, natural vegetable fibres (e.g. jute, coir) it is to be noted that normal pre-treatment with light petroleum and water does not remove all the natural non-fibrous substances; nevertheless additional pre-treatment is not applied unless the sample contains finishes insoluble in both light petroleum and water.
Analysis reports shall include full details of the methods of pre-treatment used.
I.7.TEST PROCEDUREU.K.
I.7.1.General instructionsU.K.
I.7.1.1.DryingU.K.
Conduct all drying operations for not less than 4 hours and not more than 16 hours at 105 ± 3 °C in a ventilated oven with the oven door closed throughout. If the drying period is less than 14 hours, the specimen must be checkweighed to determine whether its mass is constant. The mass may be considered as constant if, after a further drying period of 60 minutes, its variation is less than 0,05 %.
Avoid handling crucibles and weighing bottles, specimens or residues with bare hands during the drying, cooling and weighing operations.
Dry specimens in a weighing bottle with its cover beside it. After drying, stopper the weighing bottle before removing it from the oven, and transfer it quickly to the desiccator.
Dry the filter crucible in a weighing bottle with its cover beside it in the oven. After drying, close the weighing bottle and transfer it quickly to the desiccator.
Where apparatus other than a filter crucible is used, drying operations shall be conducted in the oven so as to determine the dry mass of the fibres without loss.
I.7.1.2.CoolingU.K.
Conduct all cooling operations in the desiccator, placed beside the balance, until the cooling of the weighing bottles is complete, and in any case for not less than 2 hours.
I.7.1.3.WeighingU.K.
After cooling, complete the weighing of the weighing bottle within 2 minutes of its removal from the desiccator; weigh to an accuracy of 0,0002 g.
I.7.2.ProcedureU.K.
Take from the pre-treated laboratory test sample a test specimen of at least 1 g (in mass). Cut yarn or cloth into lengths of about 10 mm, dissected as much as possible. Dry the specimen in a weighing bottle, cool it in the desiccator and weigh it. Transfer the specimen to the glass vessel specified in the appropriate section of the Union method, reweigh the weighing bottle immediately and obtain the dry mass of the specimen by difference; complete the test as specified in the appropriate section of the applicable method. Examine the residue microscopically to check that the treatment has in fact completely removed the soluble fibre(s).
I.8.CALCULATION AND EXPRESSION OF RESULTSU.K.
Express the mass of each component as a percentage of the total mass of fibre in the mixture. Calculate the results on the basis of dean dry mass, adjusted by (a) the agreed allowances and (b) the correction factors necessary to take account of loss of non-fibrous matter during pre-treatment and analysis.
I.8.1.Calculation of percentages of mass of clean dry fibres disregarding loss of fibre mass during pre-treatment.U.K.
I.8.1.1.- VARIANT 1 -U.K.
Formulae to be applied where a component of the mixture is removed from one specimen and another component from a second specimen:


P 3% = 100 – (P 1% + P 2%)
P1%
is the percentage of the first clean dry component (component in the first specimen dissolved in the first reagent),
P2%
is the percentage of the second clean dry component (component in the second specimen dissolved in the second reagent),
P3%
is the percentage of the third clean dry component (component undissolved in both specimens),
m1
is the dry mass of the first specimen after pre-treatment,
m2
is the dry mass of the second specimen after pre-treatment,
r1
is the dry mass of the residue after removal of the first component from the first specimen in the first reagent,
r2
is the dry mass of the residue after removal of the second component from the second specimen in the second reagent,
d1
is the correction factor for loss in mass in the first reagent, of the second component undissolved in the first specimen(16);
d2
is the correction factor for loss in mass in the first reagent, of the third component undissolved in the first specimen,
d3
is the correction factor for loss in mass in the second reagent, of the first component undissolved in the second specimen,
d4
is the correction factor for loss in mass in the second reagent, of the third component undissolved in the second specimen.
I.8.1.2.- VARIANT 2 -U.K.
Formulae to be applied where a component (a) is removed from the first test specimen, leaving as residue the other two components (b + c), and two components (a + b) are removed from the second test specimen, leaving as residue the third component (c):
P 1% = 100 – (P 2% + P 3%)


P1%
is the percentage of the first clean dry component (component in the first specimen dissolved in the first reagent),
P2%
is the percentage of the second clean dry component (component soluble, at the same time as the first component of the second specimen, in the second reagent),
P3%
is the percentage of the third clean dry component (component undissolved in both specimens),
m1
is the dry mass of the first specimen after pre-treatment,
m2
is the dry mass of the second specimen after pre-treatment,
r1
is the dry mass of the residue after removal of the first component from the first specimen in the first reagent,
r2
is the dry mass of the residue after removal of the first and second components from the second specimen in the second reagent,
d1
is the correction factor for loss in mass in the first reagent, of the second component undissolved in the first specimen,
d2
is the correction factor for loss in mass in the first reagent, of the third component undissolved in the first specimen,
d4
is the correction factor for loss in mass in the second reagent, of the third component undissolved in the second specimen.
I.8.1.3.- VARIANT 3 -U.K.
Formulae to be applied where two components (a + b) are removed from a specimen, leaving as residue the third component (c), then two components (b + c) are removed from another specimen, leaving as residue the first component (a):

P 2% = 100 – (P 1% + P 3%)

P1%
is the percentage of the first clean dry component (component dissolved by the reagent),
P2%
is the percentage of the second clean dry component (component dissolved by the reagent),
P3%
is the percentage of the third clean dry component (component dissolved in the second specimen by the reagent),
m1
is the dry mass of the first specimen after pre-treatment,
m2
is the dry mass of the second specimen after pre-treatment,
r1
is the dry mass of the residue after the removal of the first and second components from the first specimen with the first reagent,
r2
is the dry mass of the residue after the removal of the second and third components from the second specimen with the second reagent,
d2
is the correction factor for loss in mass in the first reagent of the third component undissolved in the first specimen,
d3
is the correction factor for loss in mass in the second reagent of the first component undissolved in the second specimen.
I.8.1.4.- VARIANT 4 -U.K.
Formulae to be applied where two components are successively removed from the mixture using the same specimen:
P 1% = 100 – (P 2% + P 3%)

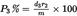
P1%
is the percentage of the first clean dry component (first soluble component),
P2%
is the percentage of the second clean dry component (second soluble component),
P3%
is the percentage of the third clean dry component (insoluble component),
m
is the dry mass of the specimen after pre-treatment,
r1
is the dry mass of the residue after elimination of the first component by the first reagent,
r2
is the dry mass of the residue after elimination of the first and second component by the first and second reagents,
d1
is the correction factor for loss in mass of the second component in the first reagent,
d2
is the correction factor for loss in mass of the third component in the first reagent,
d3
is the correction factor for loss in mass of the third component in the first and second reagents(17).
I.8.2.Calculation of the percentage of each component with adjustment by agreed allowances and, where appropriate, correction factors for losses in mass during pre-treatment operations:U.K.
Given:



then:



P1A%
is the percentage of the first clean dry component, including moisture content and loss in mass during pre-treatment,
P2A%
is the percentage of the second clean dry component, including moisture content and loss in mass during pre-treatment,
P3A%
is the percentage of the third clean dry component, including moisture content and loss in mass during pre-treatment,
P1
is the percentage of the first clean dry component obtained by one of the formulae given in I.8.1,
P2
is the percentage of the second clean dry component obtained by one of the formulae given in I.8.1,
P3
is the percentage of the third clean dry component obtained by one of the formulae given in I.8.1,
a1
is the agreed allowance of the first component,
a2
is the agreed allowance of the second component,
a3
is the agreed allowance of the third component,
b1
is the percentage of loss in mass of the first component during pre-treatment,
b2
is the percentage of loss in mass of the second component during pre-treatment,
b3
is the percentage of loss in mass of the third component during pre-treatment.
Where a special pre-treatment is used the values b1, b2 and b3 shall be determined, if possible, by submitting each of the pure fibre constituents to the pre-treatment applied in the analysis. Pure fibres are those free from all non-fibrous material except those which they normally contain (either naturally or because of the manufacturing process), in the state (unbleached, bleached) in which they are found in the material to be analysed.
Where no clean separate constituent fibres used in the manufacture of the material to be analysed are available, average values of b1, b2 and b3 as obtained from tests performed on clean fibres similar to those in the mixture under examination, must be used.
If normal pre-treatment by extraction with light petroleum and water is applied, correction factors b1, b2 and b3 may generally be ignored, except in the case of unbleached cotton, unbleached flax (or linen) and unbleached hemp where the loss due to pre-treatment is usually accepted as 4 % and in the case of polypropylene as 1 %.
In the case of other fibres, losses due to pre-treatment are usually disregarded in calculations.
I.8.3.Note:U.K.
Calculation examples are given in Section IV.
II. Method of quantitative analysis by manual separation of ternary fibre mixtures U.K.
II.1.FIELD OF APPLICATIONU.K.
This method is applicable to textile fibres of all types provided they do not form an intimate mixture and that it is possible to separate them by hand.
II.2.PRINCIPLEU.K.
After identification of the textile components, the non-fibrous matter is removed by a suitable pre-treatment and then the fibres are separated by hand, dried and weighed in order to calculate the proportion of each fibre in the mixture.
II.3.APPARATUSU.K.
II.3.1.Weighing bottles or other apparatus giving identical results.U.K.
II.3.2.Desiccator containing self-indicating silica gel.U.K.
II.3.3.Ventilated oven for drying specimens at 105 ± 3 °C.U.K.
II.3.4.Analytical balance accurate to 0,0002 g.U.K.
II.3.5.Soxhlet extractor, or other apparatus giving identical results.U.K.
II.3.6.Needle.U.K.
II.3.7.Twist tester or similar apparatus.U.K.
II.4.REAGENTSU.K.
II.4.1.Light petroleum, redistilled, boiling range 40 to 60 °C.U.K.
II.4.2.Distilled or deionised water.U.K.
II.5.CONDITIONING AND TESTING ATMOSPHEREU.K.
See I.4.
II.6.LABORATORY TEST SAMPLEU.K.
See I.5.
II.7.PRE-TREATMENT OF LABORATORY TEST SAMPLESU.K.
See I.6.
II.8.PROCEDUREU.K.
II.8.1.Analysis of yarnU.K.
Take from the pre-treated laboratory test sample a specimen of mass not less than 1 g. For a very fine yarn, the analysis may be made on a minimum length of 30 m, whatever its mass.
Cut the yarn into pieces of a suitable length and separate the fibre types by means of a needle and, if necessary, a twist tester. The fibre types so obtained are placed in pre-weighed weighing bottles and dried at 105 ± 3 °C to constant mass, as described in I.7.1 and I.7.2.
II.8.2.Analysis of clothU.K.
Take from the pre-treated laboratory test sample a specimen of mass not less than 1 g, not including a selvedge with edges carefully trimmed to avoid fraying and running parallel with weft or warp yarns, or in the case of knitted fabrics in the line of the wales and courses. Separate the different types of fibres and collect them in pre-weighed weighing bottles and proceed as described in II.8.1.
II.9.CALCULATION AND EXPRESSION OF RESULTSU.K.
Express the mass of each component fibre as a percentage of the total mass of the fibres in the mixture. Calculate the results on the basis of clean dry mass, adjusted by (a) the agreed allowances and (b) the correction factors necessary to take account of losses in mass during pre-treatment operations.
II.9.1.Calculation of percentage masses of clean dry fibre, disregarding loss in fibre mass during pre-treatment:U.K.


P 3% = 100 – (P 1% + P 2%)
P1%
is the percentage of the first clean dry component,
P2%
is the percentage of the second clean dry component,
P3%
is the percentage of the third clean dry component,
m1
is the clean dry mass of the first component,
m2
is the clean dry mass of the second component,
m3
is the clean dry mass of the third component.
II.9.2.For calculation of the percentage of each component with adjustment by agreed allowances and, where appropriate, by correction factors for losses in mass during pre-treatment: see I.8.2.U.K.
III. Method of quantitative analysis of ternary fibre mixtures by a combination of manual separation and chemical separation U.K.
Wherever possible, manual separation shall be used, taking account of the proportions of components separated before proceeding to any chemical treatment of each of the separate components.
III.1.PRECISION OF THE METHODSU.K.
The precision indicated in each method of analysis of binary fibre mixtures relates to the reproducibility (see Chapter 2 relating to methods for quantitative analysis of certain binary textile fibre mixtures).
Reproducibility refers to the reliability, i.e. the closeness of agreement between experimental values obtained by operators in different laboratories or at different times using the same method and obtaining individual results on specimens of an identical homogeneous mixture.
Reproducibility is expressed by confidence limits of the results for a confidence level of 95 %.
By this is meant that the difference between two results in a series of analyses made in different laboratories would, given a normal and correct application of the method to an identical and homogeneous mixture, exceed the confidence limit only in five cases out of 100.
To determine the precision of the analysis of a ternary fibre mixture the values indicated in the methods for the analysis of binary fibre mixtures which have been used to analyse the ternary fibre mixture are applied in the usual way.
Given that in the four variants of the quantitative chemical analysis of ternary fibre mixtures, provision is made for two dissolutions (using two separate specimens for the first three variants and a single specimen for the fourth variant) and, assuming that E1 and E2 denote the precision of the two methods for analysing binary fibre mixtures, the precision of the results for each component is shown in the following table:
| Component fibre | Variants | ||
|---|---|---|---|
| 1 | 2 and 3 | 4 | |
| a | E1 | E1 | E1 |
| b | E2 | E1 + E2 | E1 + E2 |
| c | E1 + E2 | E2 | E1 + E2 |
If the fourth variant is used, the degree of precision may be found to be lower than that calculated by the method indicated above, owing to possible action of the first reagent on the residue consisting of components b and c, which would be difficult to evaluate.
III.2.TEST REPORTU.K.
III.2.1.Indicate the variant(s) used to carry out the analysis, the methods, reagents and correction factors.U.K.
III.2.2.Give details of any special pre-treatments (see I.6).U.K.
III.2.3.Give the individual results and the arithmetic mean, each to the first decimal place.U.K.
III.2.4.Wherever possible, state the precision of the method for each component, calculated according to the table in Section III.1.U.K.
IV. Examples of the calculation of percentages of the components of certain ternary fibre mixtures using some of the variants described in point I.8.1. U.K.
Consider the case of a fibre mixture which gave the following components when qualitatively analysed for raw material composition: 1. carded wool; 2. nylon (polyamide); 3. unbleached cotton.
VARIANT No 1U.K.
Using this variant, that is using two different specimens and removing one component (a = wool) by dissolution from the first specimen and a second component (b = polyamide) from the second specimen, the following results can be obtained:
1.
Dry mass of the first specimen after pre-treatment is (m1) = 1,6000 g
2.
Dry mass of the residue after treatment with alkaline sodium hypochlorite (polyamide + cotton) (r1) = 1,4166 g
3.
Dry mass of the second specimen after pre-treatment (m2) = 1,8000 g
4.
Dry mass of the residue after treatment with formic acid (wool + cotton) (r2) = 0,9000 g
Treatment with alkaline sodium hypochlorite does not entail any loss in mass of polyamide, while unbleached cotton loses 3 %, therefore d1 = 1,00 and d2 = 1,03.
Treatment with formic acid does not entail any loss in mass for wool or unbleached cotton, therefore d3 and d4 = 1,00.
If the values obtained by chemical analysis and the correction factors are substituted in the formula under I.8.1.1, the following result is obtained:
P1% (wool) = [1,03/1,00 – 1,03 × 1,4166/1,6000 + (0,9000/1,8000) × (1 – 1,03/1,00)] ×100 = 10,30
P2% (polyamide) = [1,00/1,00 – 1,00 × 0,9000/1,8000 + (1,4166/1,6000) × (1 – 1,00/1,00)] × 100 = 50,00
P3% (cotton) = 100 – (10,30 + 50,00) = 39,70
The percentages of the various clean dry fibres in the mixture are as follows:
| wool | 10,3 % |
| polyamide | 50,0 % |
| cotton | 39,7 % |
These percentages must be corrected according to the formulae under I.8.2, in order to take account of the agreed allowances and the correction factors for any losses in mass after pre-treatment.
As indicated in Annex IX, the agreed allowances are as follows: carded wool 17,00 %, polyamide 6,25 %, cotton 8,50 %, also unbleached cotton shows a loss in mass of 4 %, after pre-treatment with light petroleum and water.
Therefore:
P1A% (wool) = 10,30 × [1 + (17,00 + 0,0)/100] / [10,30 × (1 + (17,00 + 0,0)/100) + 50,00 × (1 + (6,25 + 0,0)/100) + 39,70 × (1 + (8,50 + 4,0)/100)] × 100 = 10,97
P2A% (polyamide) = 50,0 × [(1 + (6,25 + 0,0)/100)/109,8385] × 100 = 48,37
P3A% (cotton) = 100 – (10,97 + 48,37) = 40,66
The raw material composition of the yarn is therefore as follows:
| polyamide | 48,4 % |
| cotton | 40,6 % |
| wool | 11,0 % |
| 100,0 % |
VARIANT No 4U.K.
Consider the case of a fibre mixture which when qualitatively analysed gave the following components: carded wool, viscose, unbleached cotton.
Suppose that using variant 4, that is successively removing two components from the mixture of one single specimen, the following results are obtained:
1.
Dry mass of the specimen after pre-treatment (m) = 1,6000 g
2.
Dry mass of the residue after treatment with alkaline sodium hypochlorite (viscose + cotton) (r1) = 1,4166 g
3.
Dry mass of the residue after the second treatment of the residue r1 with zinc chloride/formic acid (cotton) (r2) = 0,6630 g
Treatment with alkaline sodium hypochlorite does not entail any loss in mass of viscose, while unbleached cotton loses 3 %, therefore d1 =1,00 and d2 = 1,03.
As a result of treatment with formic acid-zinc chloride, the mass of cotton increases by 4 %, so that d3 = 1,03 × 0,96 = 0,9888, rounded to 0,99, (d3 being the correction factor for the respective loss or increase in mass of the third component in the first and second reagents).
If the values obtained by chemical analysis and the correction factors are substituted in the formulae given in I.8.1.4, the following result is obtained:
P2% (viscose) = 1,00 × (1,4166/1,6000) × 100 – (1,00/1,03) × 41,02 = 48,71 %
P3% (cotton) = 0,99 × (0,6630/1,6000) × 100 = 41,02 %
P1% (wool) = 100 – (48,71 + 41,02) = 10,27 %
As has already been indicated for Variant 1, these percentages must be corrected by the formulae indicated in point I.8.2.
P1A% (wool) = 10,27 × [1 + (17,0 + 0,0)/100)]/[10,27 × (1 + (17,00 + 0,0)/100) +48,71 × (1 + (13 + 0,0)/100) + 41,02 × (1 + (8,5 + 4,0)/100)] × 100 = 10,61 %
P2A% (viscose) = 48,71 × [1 + (13 + 0,0)/100] / 113,2057 × 100 = 48,62 %
P3A% (cotton) = 100 – (10,61 + 48,62) = 40,77 %
The raw material composition of the mixture is therefore as follows:
| viscose | 48,6 % |
| cotton | 40,8 % |
| wool | 10,6 % |
| — | |
| 100,0 % |
V. Table of typical ternary fibre mixtures which may be analysed using Union methods of analysis of binary fibre mixtures (for illustration purposes) U.K.
| Mixture No | Component fibres | Variant | Number of method used and reagent for binary fibre mixtures | ||
|---|---|---|---|---|---|
| Component 1 | Component 2 | Component 3 | |||
| 1. | wool or hair | viscose, cupro or certain types of modal | cotton | 1 and/or 4 | 2. (hypochlorite) and 3. (zinc chloride/formic acid) |
| 2. | wool or hair | polyamide or nylon | cotton, viscose, cupro or modal | 1 and/or 4 | 2. (hypochlorite) and 4. (formic acid, 80 % m/m) |
| 3. | wool, hair or silk | certain other fibres | viscose, cupro modal or cotton | 1 and/or 4 | 2. (hypochlorite) and 9. (carbon disulphide/acetone 55,5/44,5 % v/v) |
| 4. | wool or hair | polyamide or nylon | polyester, polypropylene, acrylic or glass fibre | 1 and/or 4 | 2. (hypochlorite) and 4. (formic acid, 80 % m/m) |
| 5. | wool, hair or silk | certain other fibres | polyester, acrylic, polyamide or nylon or glass fibre | 1 and/or 4 | 2. (hypochlorite) and 9. (carbon disulphide/acetone 55,5/44,5 % v/v) |
| 6. | silk | wool or hair | polyester | 2 | 11. (sulphuric acid 75 % m/m) and 2. (hypochlorite) |
| 7. | polyamide or nylon | acrylic or certain other fibres | cotton, viscose, cupro or modal | 1 and/or 4 | 4. (formic acid 80 % m/m) and 8. (dimethylformamide) |
| 8. | certain chlorofibres | polyamide or nylon | cotton, viscose, cupro or modal | 1 and/or 4 | 8. (dimethylformamide) and 4. (formic acid, 80 % m/m) or 9. (carbon disulphide/acetone, 55,5/44,5 % v/v) and 4. (formic acid, 80 % m/m) |
| 9. | acrylic | polyamide or nylon | polyester | 1 and/or 4 | 8. (dimethylformamide) and 4. (formic acid, 80 % m/m) |
| 10. | acetate | polyamide or nylon or certain other fibres | viscose, cotton, cupro or modal | 4 | 1. (acetone) and 4. (formic acid, 80 % m/m) |
| 11. | certain chlorofibres | acrylic or certain other fibres | polyamide or nylon | 2 and/or 4 | 9. (carbon disulphide/acetone 55,5/44,5 % v/v) and 8. (dimethylformamide) |
| 12. | certain chlorofibres | polyamide or nylon | acrylic | 1 and/or 4 | 9. (carbon disulphide/acetone 55,5/44,5 % v/v) and 4. (formic acid, 80 %m/m) |
| 13. | polyamide or nylon | viscose, cupro, modal or cotton | polyester | 4 | 4. (formic acid, 80 % m/m) and 7. (sulphuric acid, 75 % m/m) |
| 14. | acetate | viscose, cupro, modal or cotton | polyester | 4 | 1. (acetone) and 7 (sulphuric acid, 75 % m/m) |
| 15. | acrylic | viscose, cupro, modal or cotton | polyester | 4 | 8. (dimethylformamide) and 7. (sulphuric acid, 75 % m/m) |
| 16. | acetate | wool, hair or silk | cotton, viscose, cupro, modal, polyamide or nylon, polyester, acrylic | 4 | 1. (acetone) and 2. (hypochlorite) |
| 17. | triacetate | wool, hair or silk | cotton, viscose, cupro, modal, polyamide or nylon, polyester, acrylic | 4 | 6. (dichloromethane) and 2. (hypochlorite) |
| 18. | acrylic | wool, hair or silk | polyester | 1 and/or 4 | 8. (dimethylformamide) and 2. (hypochlorite) |
| 19. | acrylic | silk | wool or hair | 4 | 8. (dimethylformamide) and 11. (sulphuric acid 75 % m/m) |
| 20. | acrylic | wool or hair silk | cotton, viscose, cupro or modal | 1 and/or 4 | 8. (dimethylformamide) and 2. (hypochlorite) |
| 21. | wool, hair or silk | cotton, viscose, modal, cupro | polyester | 4 | 2. (hypochlorite) and 7. (sulphuric acid 75 % m/m) |
| 22. | viscose, cupro or certain types of modal | cotton | polyester | 2 and/or 4 | 3. (zinc chloride/formic acid) and 7. (sulphuric acid 75 % m/m) |
| 23. | acrylic | viscose, cupro or certain types of modal | cotton | 4 | 8. (dimethylformamide) and 3 (zinc chloride/formic acid) |
| 24. | certain chlorofibres | viscose, cupro or certain types of modal | cotton | 1 and/or 4 | 9. (carbon disulphide/acetone, 55,5/44,5 % v/v) and 3. (zinc chloride/formic acid) or 8. (dimethylformamide) and 3. (zinc chloride/formic acid) |
| 25. | acetate | viscose, cupro or certain types of modal | cotton | 4 | 1. (acetone) and 3. (zinc chloride/formic acid) |
| 26. | triacetate | viscose, cupro or certain types of modal | cotton | 4 | 6. (dichloromethane) and 3. (zinc chloride/formic acid) |
| 27. | acetate | silk | wool or hair | 4 | 1. (acetone) and 11. (sulphuric acid 75 % m/m) |
| 28. | triacetate | silk | wool or hair | 4 | 6. (dichloromethane) and 11. (sulphuric acid 75 % m/m) |
| 29. | acetate | acrylic | cotton, viscose, cupro or modal | 4 | 1. (acetone) and 8. (dimethylformamide) |
| 30. | triacetate | acrylic | cotton, viscose, cupro or modal | 4 | 6. (dichloromethane) and 8. (dimethylformamide) |
| 31. | triacetate | polyamide or nylon | cotton, viscose, cupro or modal | 4 | 6. (dichloromethane) and 4. (formic acid 80 % m/m) |
| 32. | triacetate | cotton, viscose, cupro or modal | polyester | 4 | 6. (dichloromethane) and 7. (sulphuric acid 75 % m/m) |
| 33. | acetate | polyamide or nylon | polyester or acrylic | 4 | 1. (acetone) and 4. (formic acid 80 % m/m) |
| 34. | acetate | acrylic | polyester | 4 | 1. (acetone) and 8. (dimethylformamide) |
| 35. | certain chlorofibres | cotton, viscose, cupro or modal | polyester | 4 | 8. (dimethylformamide) and 7. (sulphuric acid 75 % m/m) or 9 (carbon disulphide/acetone, 55,5/44,5 % v/v) and 7. (sulphuric acid 75 % m/m) |
| 36. | cotton | polyester | elastolefin | 2 and/or 4 | 7. (sulphuric acid 75 % m/m) and 14. (concentrated sulphuric acid) |
| 37. | certain modacrylics | polyester | melamine | 2 and/or 4 | 8. (dimethylformamide) and 14. (concentrated sulphuric acid) |
ANNEX IXU.K.
Agreed allowances used to calculate the mass of fibres contained in a textile product
(referred to in Article 19(3))
| a The agreed allowances of 17,00 % shall also be applied where it is impossible to ascertain whether the textile product containing wool and/or animal hair is combed or carded. | ||
| Fibre No | Fibres | Percentages |
|---|---|---|
| 1-2 | Wool and animal hair: | |
| combed fibres | 18,25 | |
| carded fibres | 17,0a | |
| 3 | Animal hair: | |
| combed fibres | 18,25 | |
| carded fibres | 17,0a | |
| Horsehair: | ||
| combed fibres | 16,0 | |
| carded fibres | 15,0 | |
| 4 | Silk | 11,0 |
| 5 | Cotton: | |
| normal fibres | 8,5 | |
| mercerised fibres | 10,5 | |
| 6 | Kapok | 10,9 |
| 7 | Flax (or linen) | 12,0 |
| 8 | True hemp | 12,0 |
| 9 | Jute | 17,0 |
| 10 | Abaca | 14,0 |
| 11 | Alfa | 14,0 |
| 12 | Coir | 13,0 |
| 13 | Broom | 14,0 |
| 14 | Ramie (bleached fibre) | 8,5 |
| 15 | Sisal | 14,0 |
| 16 | Sunn | 12,0 |
| 17 | Henequen | 14,0 |
| 18 | Maguey | 14,0 |
| 19 | Acetate | 9,0 |
| 20 | Alginate | 20,0 |
| 21 | Cupro | 13,0 |
| 22 | Modal | 13,0 |
| 23 | Protein | 17,0 |
| 24 | Triacetate | 7,0 |
| 25 | Viscose | 13,0 |
| 26 | Acrylic | 2,0 |
| 27 | Chlorofibre | 2,0 |
| 28 | Fluorofibre | 0,0 |
| 29 | Modacrylic | 2,0 |
| 30 | Polyamide or nylon: | |
| discontinuous fibre | 6,25 | |
| filament | 5,75 | |
| 31 | Aramid | 8,0 |
| 32 | Polyimide | 3,5 |
| 33 | Lyocell | 13,0 |
| 34 | Polylactide | 1,5 |
| 35 | Polyester | 1,5 |
| 36 | Polyethylene | 1,5 |
| 37 | Polypropylene | 2,0 |
| 38 | Polycarbamide | 2,0 |
| 39 | Polyurethane: | |
| discontinuous fibre | 3,5 | |
| filament | 3,0 | |
| 40 | Vinylal | 5,0 |
| 41 | Trivinyl | 3,0 |
| 42 | Elastodiene | 1,0 |
| 43 | Elastane | 1,5 |
| 44 | Glass fibre: | |
| with an average diameter of over 5 μm | 2,0 | |
| with an average diameter of 5 μm or less | 3,0 | |
| 45 | Elastomultiester | 1,5 |
| 46 | Elastolefin | 1,5 |
| 47 | Melamine | 7,0 |
| 48 | Metal fibre | 2,0 |
| Metallised fibre | 2,0 | |
| Asbestos | 2,0 | |
| Paper yarn | 13,75 | |
| [F149 | Polypropylene/polyamide bicomponent | 1,0] |
| [F250 | Polyacrylate | 30,0] |
ANNEX XU.K.
Correlation Tables
| Directive 2008/121/EC | This Regulation |
|---|---|
| Article 1(1) | Article 4 |
| Article 1(2)(a)-(c) | — |
| Article 1(2)(d) | Article 2(3) |
| Article 2(1) | Article 3(1) |
| Article 2(2) introductory wording | Article 2(2) introductory wording |
| Article 2(2)(a) | Article 2(2)(a) |
| Article 2(2)(b) | Article 2(2)(b) and (c) |
| Article 2(2)(c) | Article 2(2)(d) |
| Article 3 | Article 5 |
| Article 4 | Article 7 |
| Article 5 | Article 8 |
| Article 6(1) and (2) | — |
| Article 6(3) | Article 9(3) |
| Article 6(4) | Article 9(4) |
| Article 6(5) | Article 20 |
| Article 7 | Article 10 |
| Article 8(1) first sentence | Article 14(1) |
| Article 8(1) second sentence | Article 14(2) |
| Article 8(2) | Article 14(3) |
| Article 8(3) first subparagraph | Article 16(1) |
| Article 8(3) second and third subparagraph | Article 16(2) |
| Article 8(4) | Article 16(3) |
| Article 8(5) | — |
| Article 9(1) | Article 11(1) and (2) |
| Article 9(2) | Article 11(3) |
| Article 9(3) | Article 13 and Annex IV |
| Article 10(1)(a) | Article 17(2) |
| Article 10(1)(b) | Article 17(3) |
| Article 10(1)(c) | Article 17(4) |
| Article 10(2) | Article 17(5) |
| Article 11 | Article 15(4) |
| Article 12 | Article 19(2) and Annex VII |
| Article 13(1) | Article 19(1) |
| Article 13(2) | — |
| Article 14(1) | — |
| Article 14(2) | — |
| Article 15 | Article 21 |
| Article 16 | — |
| Article 17 | — |
| Article 18 | — |
| Article 19 | — |
| Article 20 | — |
| Annex I | Annex I |
| Annex II | Annex III |
| Annex III | Annex V |
| Annex III point 36 | Article 3(1)(j) |
| Annex IV | Annex VI |
| Annex V | Annex IX |
| Annex VI | — |
| Annex VII | — |
| Directive 96/73/EC | This Regulation |
|---|---|
| Article 1 | Article 1 |
| Article 2 | Annex VIII Chapter 1 Section I (2) |
| Article 3 | Article 19(1) |
| Article 4 | Article 19(4) |
| Article 5 | Article 21 |
| Article 6 | — |
| Article 7 | — |
| Article 8 | — |
| Article 9 | — |
| Annex I | Annex VIII Chapter 1 Section I |
| Annex II | Annex VIII Chapter 1 Section II and Chapter 2 |
| Annex III | — |
| Annex IV | — |
| Directive 73/44/EEC | This Regulation |
|---|---|
| Article 1 | Article 1 |
| Article 2 | Annex VIII Chapter 1 Section I |
| Article 3 | Article 19(1) |
| Article 4 | Article 19(4) |
| Article 5 | Article 21 |
| Article 6 | — |
| Article 7 | — |
| Annex I | Annex VIII Chapter 3 introduction and Sections I to III |
| Annex II | Annex VIII Chapter 3 Section IV |
| Annex III | Annex VIII Chapter 3 Section V |
(1)
For the products falling within this item and sold in cut lengths, the inclusive labelling shall be that of the reel. The cordage and ropes falling within this item include those used in mountaineering and water sports.
(2)
In some cases it is necessary to pre-treat the individual test specimen.
(3)
For made-up and finished articles see point 7.
(4)
See point 1.
(5)
The laboratory carder may be replaced by a fibre blender, or the fibres may be mixed by the method of ‘tufts and rejects’.
(6)
If the packages can be mounted in a convenient creel a number can be wound simultaneously.
(7)
Method 12 is an exception. It is based on a determination of the content of a constituent substance of one of the two components.
(8)
See Chapter 1.1.
(9)
To ensure that the fibrous residue is immersed in the ammonia solution for 10 minutes, one may, for example, use a filter crucible adaptor fitted with a tap by which the flow of the ammonia solution can be regulated.
(10)
The solubility of such modacrylics or chlorofibres in the reagent shall be checked before carrying out the analysis.
(11)
Before carrying out the analysis, the solubility of the polyvinyl chloride fibres in the reagent shall be checked.
(12)
[F5Wild silks, such as tussah silk, are not completely soluble in 75 % m/m sulphuric acid.]
(13)
These reagents should be nitrogen-free.
(14)
See for example the apparatus described in Melliand Textilberichte 56 (1975), pp. 643-645.
(15)
See Chapter 1.1.
(16)
The values of d are indicated in Chapter 2 of this Annex relating to the various methods of analysing binary mixtures.
(17)
Wherever possible d3 should be determined in advance by experimental methods.
Textual Amendments
F5 Substituted by Commission Delegated Regulation (EU) No 286/2012 of 27 January 2012 amending, in order to include a new textile fibre name, Annex I, and, for the purposes of their adaptation to technical progress, Annexes VIII and IX to Regulation (EU) No 1007/2011 of the European Parliament and of the Council on textile fibre names and related labelling and marking of the fibre composition of textile products (Text with EEA relevance).
Options/Help
Print Options
PrintThe Whole Regulation
PrintThe Annexes only
You have chosen to open the Whole Regulation
The Whole Regulation you have selected contains over 200 provisions and might take some time to download. You may also experience some issues with your browser, such as an alert box that a script is taking a long time to run.
Would you like to continue?
You have chosen to open Schedules only
The Schedules you have selected contains over 200 provisions and might take some time to download. You may also experience some issues with your browser, such as an alert box that a script is taking a long time to run.
Would you like to continue?
Legislation is available in different versions:
Latest Available (revised):The latest available updated version of the legislation incorporating changes made by subsequent legislation and applied by our editorial team. Changes we have not yet applied to the text, can be found in the ‘Changes to Legislation’ area.
Original (As adopted by EU): The original version of the legislation as it stood when it was first adopted in the EU. No changes have been applied to the text.
Point in Time: This becomes available after navigating to view revised legislation as it stood at a certain point in time via Advanced Features > Show Timeline of Changes or via a point in time advanced search.
See additional information alongside the content
Geographical Extent: Indicates the geographical area that this provision applies to. For further information see ‘Frequently Asked Questions’.
Show Timeline of Changes: See how this legislation has or could change over time. Turning this feature on will show extra navigation options to go to these specific points in time. Return to the latest available version by using the controls above in the What Version box.
Opening Options
Different options to open legislation in order to view more content on screen at once
More Resources
Access essential accompanying documents and information for this legislation item from this tab. Dependent on the legislation item being viewed this may include:
- the original print PDF of the as adopted version that was used for the EU Official Journal
- lists of changes made by and/or affecting this legislation item
- all formats of all associated documents
- correction slips
- links to related legislation and further information resources
Timeline of Changes
This timeline shows the different versions taken from EUR-Lex before exit day and during the implementation period as well as any subsequent versions created after the implementation period as a result of changes made by UK legislation.
The dates for the EU versions are taken from the document dates on EUR-Lex and may not always coincide with when the changes came into force for the document.
For any versions created after the implementation period as a result of changes made by UK legislation the date will coincide with the earliest date on which the change (e.g an insertion, a repeal or a substitution) that was applied came into force. For further information see our guide to revised legislation on Understanding Legislation.
More Resources
Use this menu to access essential accompanying documents and information for this legislation item. Dependent on the legislation item being viewed this may include:
- the original print PDF of the as adopted version that was used for the print copy
- correction slips
Click 'View More' or select 'More Resources' tab for additional information including:
- lists of changes made by and/or affecting this legislation item
- confers power and blanket amendment details
- all formats of all associated documents
- links to related legislation and further information resources
 All content is available under the Open Government Licence v3.0 except where otherwise stated. This site additionally contains content derived from EUR-Lex, reused under the terms of the Commission Decision 2011/833/EU on the reuse of documents from the EU institutions. For more information see the EUR-Lex public statement on re-use.
All content is available under the Open Government Licence v3.0 except where otherwise stated. This site additionally contains content derived from EUR-Lex, reused under the terms of the Commission Decision 2011/833/EU on the reuse of documents from the EU institutions. For more information see the EUR-Lex public statement on re-use.