- Latest available (Revised)
- Original (As adopted by EU)
Commission Regulation (EU) 2017/735Show full title
Commission Regulation (EU) 2017/735 of 14 February 2017 amending, for the purpose of its adaptation to technical progress, the Annex to Regulation (EC) No 440/2008 laying down test methods pursuant to Regulation (EC) No 1907/2006 of the European Parliament and of the Council on the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH) (Text with EEA relevance)
You are here:
- Regulations originating from the EU
- 2017 No. 735
- ANNEX
- Division B.60

What Version
More Resources
Legislation originating from the EU
When the UK left the EU, legislation.gov.uk published EU legislation that had been published by the EU up to IP completion day (31 December 2020 11.00 p.m.). On legislation.gov.uk, these items of legislation are kept up-to-date with any amendments made by the UK since then.


This item of legislation originated from the EU
Legislation.gov.uk publishes the UK version. EUR-Lex publishes the EU version. The EU Exit Web Archive holds a snapshot of EUR-Lex’s version from IP completion day (31 December 2020 11.00 p.m.).
Status:
This is the original version as it was originally adopted in the EU.
This legislation may since have been updated - see the latest available (revised) version
B.60 In Vitro Skin Sensitisation: ARE-Nrf2 Luciferase Test Method
INTRODUCTION
This test method (TM) is equivalent to OECD test guideline (TG) 442D (2015). A skin sensitiser refers to a substance that will lead to an allergic response following skin contact as defined by the United Nations Globally Harmonized System of Classification and Labelling of Chemicals (UN GHS) (1) and Regulation (EC) No 1272/2008 of the European Parliament and of the Council on Classification, Labelling and Packaging of Substances and Mixtures (CLP)(1). This test method provides an in vitro procedure (the ARE-Nrf2 luciferase assay) to be used for supporting the discrimination between skin sensitisers and non-sensitisers in accordance with the UN GHS (1) and CLP.
There is general agreement regarding the key biological events underlying skin sensitisation. The existing knowledge of the chemical and biological mechanisms associated with skin sensitisation has been summarised in the form of an Adverse Outcome Pathway (AOP) (2), going from the molecular initiating event through the intermediate events up to the adverse health effect, i.e. allergic contact dermatitis in humans or contact hypersensitivity in rodents (2) (3). The molecular initiating event is the covalent binding of electrophilic substances to nucleophilic centres in skin proteins. The second key event in this AOP takes place in the keratinocytes and includes inflammatory responses as well as gene expression associated with specific cell signalling pathways such as the antioxidant/electrophile response element (ARE)-dependent pathways. The third key event is the activation of dendritic cells, typically assessed by expression of specific cell surface markers, chemokines and cytokines. The fourth key event is T-cell proliferation, which is indirectly assessed in the murine Local Lymph Node Assay (4).
The assessment of skin sensitisation has typically involved the use of laboratory animals. The classical methods based on guinea-pigs, the Magnusson Kligman Guinea Pig Maximisation Test (GMPT) and the Buehler Test (TM B.6 (5)), study both the induction and elicitation phases of skin sensitisation. A murine test, the Local Lymph Node Assay (LLNA) (TM B.42 (4)) and its two non-radioactive modifications, LLNA: DA (TM B.50 (6)) and LLNA: BrdU-ELISA (TM B.51 (7)), which all assess the induction response exclusively, have also gained acceptance since they provide advantages over the guinea pig tests in terms of both animal welfare and objective measurement of the induction phase of skin sensitisation.
More recently, mechanistically-based in chemico and in vitro test methods have been considered scientifically valid for the evaluation of the skin sensitisation hazard of chemicals. However, combinations of non-animal methods (in silico, in chemico, in vitro) within Integrated Approaches to Testing and Assessment (IATA) will be needed to be able to fully substitute for the animal tests currently in use given the restricted AOP mechanistic coverage of each of the currently available non-animal test methods (2) (3).
This test method (ARE-Nrf2 luciferase assay) is proposed to address the second key event as explained in paragraph 2. Skin sensitisers have been reported to induce genes that are regulated by the antioxidant response element (ARE) (8) (9). Small electrophilic substances such as skin sensitisers can act on the sensor protein Keap1 (Kelch-like ECH-associated protein 1), by e.g. covalent modification of its cysteine residue, resulting in its dissociation from the transcription factor Nrf2 (nuclear factor-erythroid 2-related factor 2). The dissociated Nrf2 can then activate ARE-dependent genes such as those coding for phase II detoxifying enzymes (8) (10) (11).
Currently, the only in vitro ARE-Nrf2 luciferase assay covered by this test method is the KeratinoSensTM assay for which validation studies have been completed (9) (12) (13) followed by an independent peer review conducted by the European Union Reference Laboratory for Alternatives to Animal Testing (EURL ECVAM) (14). The KeratinoSensTM assay was considered scientifically valid to be used as part of an IATA, to support the discrimination between skin sensitisers and non-sensitisers for the purpose of hazard classification and labelling (14). Laboratories willing to implement the test method can obtain the recombinant cell line used in the KeratinoSensTM assay by establishing a licence agreement with the test method developer (15).
Definitions are provided in Appendix 1.
INITIAL CONSIDERATIONS, APPLICABILITY AND LIMITATIONS
Since activation of the Keap1-Nrf2-ARE pathway addresses only the second key event of the skin sensitisation AOP, information from test methods based on the activation of this pathway is unlikely to be sufficient when used on its own to conclude on the skin sensitisation potential of chemicals. Therefore, data generated with the present test method should be considered in the context of integrated approaches, such as IATA, combining them with other complementary information e.g. derived from in vitro assays addressing other key events of the skin sensitisation AOP as well as non-testing methods including read-across from chemical analogues. Examples on how to use the ARE-Nrf2 luciferase test method in combination with other information are reported in literature (13) (16) (17) (18) (19).
This test method can be used to support the discrimination between skin sensitisers (i.e. UN GHS/CLP Category 1) and non-sensitisers in the context of IATA. This test method cannot be used on its own, neither to sub-categorise skin sensitisers into subcategories 1A and 1B as defined by the UN GHS/CLP nor to predict potency for safety assessment decisions. However, depending on the regulatory framework, a positive result may be used on its own to classify a chemical into UN GHS/CLP category 1.
Based on the dataset from the validation study and in-house testing used for the independent peer-review of the test method, the KeratinoSensTM assay proved to be transferable to laboratories experienced in cell culture. The level of reproducibility in predictions that can be expected from the test method is in the order of 85 % within and between laboratories (14). The accuracy (77 % - 155/201), sensitivity (78 % - 71/91) and specificity (76 % - 84/110) of the KeratinoSensTM assay for discriminating skin sensitisers (i.e. UN GHS/CLP Cat. 1) from non-sensitisers when compared to LLNA results were calculated by considering all of the data submitted to EURL ECVAM for evaluation and peer-review of the test method (14). These figures are similar to those recently published based on in-house testing of about 145 substances (77 % accuracy, 79 % sensitivity, 72 % specificity) (13). The KeratinoSensTM assay is more likely to under predict chemicals showing a low to moderate skin sensitisation potency (i.e. UN GHS/CLP subcategory 1B) than chemicals showing a high skin sensitisation potency (i.e. UN GHS/CLP subcategory 1A) (13) (14). Taken together, this information indicates the usefulness of the KeratinoSensTM assay to contribute to the identification of skin sensitisation hazard. However, the accuracy values given here for the KeratinoSensTM assay as a stand-alone test method are only indicative since the test method should be considered in combination with other sources of information in the context of an IATA and in accordance with the provisions of paragraph 9 above. Furthermore when evaluating non-animal methods for skin sensitisation, it should be kept in mind that the LLNA as well as other animal tests, may not fully reflect the situation in the species of interest i.e. humans.
The term “test chemical” is used in this test method to refer to what is being tested and is not related to the applicability of the ARE-Nrf2 luciferase test method to the testing of substances and/or mixtures. On the basis of the current data available the KeratinoSensTM assay was shown to be applicable to test chemicals covering a variety of organic functional groups, reaction mechanisms, skin sensitisation potency (as determined with in vivo studies) and physico-chemical properties (9) (12) (13) (14). Mainly mono-constituent substances were tested, although a limited amount of data also exist on the testing of mixtures (20). The test method is nevertheless technically applicable to the testing of multi-constituent substances and mixtures. However, before use of this test method on a mixture for generating data for an intended regulatory purpose, it should be considered whether, and if so why, it may provide adequate results for that purpose. Such considerations are not needed, when there is a regulatory requirement for testing of the mixture. Moreover, when testing multi-constituent substances or mixtures, consideration should be given to possible interference of cytotoxic constituents with the observed responses. The test method is applicable to test chemicals soluble or that form a stable dispersion (i.e. a colloid or suspension in which the test chemical does not settle or separate from the solvent into different phases) either in water or DMSO (including all of the test chemical components in the case of testing a multi-constituent substance or a mixture). Test chemicals that do not fulfil these conditions at the highest final required concentration of 2 000 μM (cf. paragraph 22) may still be tested at lower concentrations. In such a case, results fulfilling the criteria for positivity described in paragraph 39 could still be used to support the identification of the test chemical as a skin sensitiser, whereas a negative result obtained with concentrations < 1 000 μM should be considered as inconclusive (see prediction model in paragraph 39). In general substances with a LogP of up to 5 have been successfully tested whereas extremely hydrophobic substances with a LogP above 7 are outside the known applicability of the test method (14). For substances having a LogP falling between 5 and 7, only limited information is available.
Negative results should be interpreted with caution as substances with an exclusive reactivity towards lysine-residues can be detected as negative by the test method. Furthermore, because of the limited metabolic capability of the cell line used (21) and because of the experimental conditions, pro-haptens (i.e. chemicals requiring enzymatic activation for example via P450 enzymes) and pre-haptens (i.e. chemicals activated by auto-oxidation) in particular with a slow oxidation rate may also provide negative results. Test chemicals that do not act as a sensitiser but are nevertheless chemical stressors may lead on the other hand to false positive results (14). Furthermore, highly cytotoxic test chemicals cannot always be reliably assessed. Finally, test chemicals that interfere with the luciferase enzyme can confound the activity of luciferase in cell-based assays causing either apparent inhibition or increased luminescence (22). For example, phytoestrogen concentrations higher than 1 μM were reported to interfere with the luminescence signals in other luciferase-based reporter gene assays due to over-activation of the luciferase reporter gene (23). As a consequence, luciferase expression obtained at high concentrations of phytoestrogens or similar chemicals suspected of producing phytoestrogen-like over-activation of the luciferase reporter gene needs to be examined carefully (23). In cases where evidence can be demonstrated on the non-applicability of the test method to other specific categories of test chemicals, the test method should not be used for those specific categories.
In addition to supporting discrimination between skin sensitisers and non-sensitisers, the KeratinoSensTM assay also provides concentration-response information that may potentially contribute to the assessment of sensitising potency when used in integrated approaches such as IATA (19). However, further work preferably based on reliable human data is required to determine how KeratinoSensTM assay results can contribute to potency assessment (24) and to sub-categorisation of sensitisers according to UN GHS/CLP.
PRINCIPLE OF THE TEST
The ARE-Nrf2 luciferase test method makes use of an immortalised adherent cell line derived from HaCaT human keratinocytes stably transfected with a selectable plasmid. The cell line contains the luciferase gene under the transcriptional control of a constitutive promoter fused with an ARE element from a gene that is known to be up-regulated by contact sensitisers (25) (26). The luciferase signal reflects the activation by sensitisers of endogenous Nrf2 dependent genes, and the dependence of the luciferase signal in the recombinant cell line on Nrf2 has been demonstrated (27). This allows quantitative measurement (by luminescence detection) of luciferase gene induction, using well established light producing luciferase substrates, as an indicator of the activity of the Nrf2 transcription factor in cells following exposure to electrophilic substances.
Test chemicals are considered positive in the KeratinoSens™ assay if they induce a statistically significant induction of the luciferase activity above a given threshold (i.e. > 1,5 fold or 50 % increase), below a defined concentration which does not significantly affect cell viability (i.e. below 1 000 μM and at a concentration at which the cellular viability is above 70 % (9) (12)). For this purpose, the maximal fold induction of the luciferase activity over solvent (negative) control (Imax) is determined. Furthermore, since cells are exposed to series of concentrations of the test chemicals, the concentration needed for a statistically significant induction of luciferase activity above the threshold (i.e. EC1,5 value) should be interpolated from the dose-response curve (see paragraph 32 for calculations). Finally, parallel cytotoxicity measurements should be conducted to assess whether luciferase activity induction levels occur at sub-cytotoxic concentrations.
Prior to routine use of the ARE-Nrf2 luciferase assay that adheres to this test method, laboratories should demonstrate technical proficiency, using the ten Proficiency Substances listed in Appendix 2.
Performance standards (PS) (28) are available to facilitate the validation of new or modified in vitro ARE-Nrf2 luciferase test methods similar to the KeratinoSens™ assay and allow for timely amendment of this test method for their inclusion. Mutual Acceptance of Data (MAD) according to the OECD agreement will only be guaranteed for test methods validated according to the PS, if these test methods have been reviewed and included in the corresponding test guideline by OECD.
PROCEDURE
Currently, the only method covered by this test method is the scientifically valid KeratinoSensTM assay (9) (12) (13) (14). The Standard Operating Procedures (SOP) for the KeratinoSensTM assay is available and should be employed when implementing and using the test method in the laboratory (15). Laboratories willing to implement the test method can obtain the recombinant cell line used in the KeratinoSensTM assay by establishing a licence agreement with the test method developer. The following paragraphs provide with a description of the main components and procedures of the ARE-Nrf2 luciferase test method.
Preparation of the keratinocyte cultures
A transgenic cell line having a stable insertion of the luciferase reporter gene under the control of the ARE-element should be used (e.g. the KeratinoSens™ cell line). Upon receipt, cells are propagated (e.g. 2 to 4 passages) and stored frozen as a homogeneous stock. Cells from this original stock can be propagated up to a maximum passage number (i.e. 25 in the case of KeratinoSensTM) and are employed for routine testing using the appropriate maintenance medium (in the case of KeratinoSensTM this represents DMEM containing serum and Geneticin).
For testing, cells should be 80-90 % confluent, and care should be taken to ensure that cells are never grown to full confluence. One day prior to testing cells are harvested, and distributed into 96-well plates (10 000 cells/well in the case of KeratinoSensTM). Attention should be paid to avoid sedimentation of the cells during seeding to ensure homogeneous cell number distribution across wells. If this is not the case, this step may give raise to high well-to-well variability. For each repetition, three replicates are used for the luciferase activity measurements, and one parallel replicate used for the cell viability assay.
Preparation of the test chemical and control substances
The test chemical and control substances are prepared on the day of testing. For the KeratinoSensTM assay, test chemicals are dissolved in dimethyl sulfoxide (DMSO) to the final desired concentration (e.g. 200 mM). The DMSO solutions can be considered self-sterilising, so that no sterile filtration is needed. Test chemical not soluble in DMSO is dissolved in sterile water or culture medium, and the solutions sterilised by e.g. filtration. For a test chemical which has no defined molecular weight (MW), a stock solution is prepared to a default concentration (40 mg/mL or 4 % (w/v)) in the KeratinoSensTM assay. In case solvents other than DMSO, water or the culture medium are used, sufficient scientific rationale should be provided.
Based on the stock DMSO solutions of the test chemical, serial dilutions are made using DMSO to obtain 12 master concentrations of the chemical to be tested (from 0,098 to 200 mM in the KeratinoSensTM assay). For a test chemical not soluble in DMSO, the dilutions to obtain the master concentrations are made using sterile water or sterile culture medium. Independent of the solvent used, the master concentrations, are then further diluted 25 fold into culture medium containing serum, and finally used for treatment with a further 4 fold dilution factor so that the final concentrations of the tested chemical range from 0,98 to 2 000 μM in the KeratinoSensTM assay. Alternative concentrations may be used upon justification (e.g. in case of cytotoxicity or poor solubility).
The negative (solvent) control used in the KeratinoSensTM assay is DMSO (CAS No. 67-68-5, ≥ 99 % purity), for which six wells per plate are prepared. It undergoes the same dilution as described for the master concentrations in paragraph 22, so that the final negative (solvent) control concentration is 1 %, known not to affect cell viability and corresponding to the same concentration of DMSO found in the tested chemical and in the positive control. For a test chemical not soluble in DMSO, for which the dilutions were made in water, the DMSO level in all wells of the final test solution must be adjusted to 1 % as for the other test chemicals and control substances.
The positive control used in the case of the KeratinoSensTM assay is cinnamic aldehyde (CAS No. 14371-10-9, ≥ 98 % purity), for which a series of 5 master concentrations ranging from 0,4 to 6,4 mM are prepared in DMSO (from a 6,4 mM stock solution) and diluted as described for the master concentrations in paragraph 22, so that the final concentration of the positive control range from 4 to 64 μM. Other suitable positive controls, preferentially providing EC1,5 values in the mid-range, may be used if historical data are available to derive comparable run acceptance criteria.
Application of the test chemical and control substances
For each test chemical and positive control substance, one experiment is needed to derive a prediction (positive or negative), consisting of at least two independent repetitions containing each three replicates (i.e. n = 6). In case of discordant results between the two independent repetitions, a third repetition containing three replicates should be performed (i.e. n = 9). Each independent repetition is performed on a different day with fresh stock solution of test chemicals and independently harvested cells. Cells may come from the same passage however.
After seeding as described in paragraph 20, cells are grown for 24 hours in the 96-wells microtiter plates. The medium is then removed and replaced with fresh culture medium (150 μl culture medium containing serum but without Geneticin in the case of KeratinoSensTM) to which 50 μl of the 25 fold diluted test chemical and control substances are added. At least one well per plate should be left empty (no cells and no treatment) to assess background values.
The treated plates are then incubated for about 48 hours at 37 ± 1 °C in the presence of 5 % CO2 in the KeratinoSensTM assay. Care should be taken to avoid evaporation of volatile test chemicals and cross-contamination between wells by test chemicals by e.g. covering the plates with a foil prior to the incubation with the test chemicals.
Luciferase activity measurements
Three factors are critical to ensure appropriate luminescence readings:
the choice of a sensitive luminometer,
the use of a plate format with sufficient height to avoid light-cross-contamination; and
the use of a luciferase substrate with sufficient light output to ensure sufficient sensitivity and low variability.
Prior to testing, a control experiment setup as described in Appendix 3 should be carried out to ensure that these three points are met.
After the 48 hour exposure time with the test chemical and control substances in the KeratinoSensTM assay, cells are washed with a phosphate buffered saline, and the relevant lysis buffer for luminescence readings added to each well for 20 min at room temperature.
Plates with the cell lysate are then placed in the luminometer for reading which in the KeratinoSensTM assay is programmed to: (i) add the luciferase substrate to each well (i.e. 50 μl), (ii) wait for 1 second, and (iii) integrate the luciferase activity for 2 seconds. In case alternative settings are used, e.g. depending on the model of luminometer used, these should be justified. Furthermore, a glow substrate may also be used provided that the quality control experiment of Appendix 3 is successfully fulfilled.
Cytotoxicity Assessment
For the KeratinoSensTM cell viability assay, medium is replaced after the 48 hour exposure time with fresh medium containing MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, Thiazolyl blue tetrazolium bromide; CAS No. 298-93-1) and cells incubated for 4 hours at 37 °C in the presence of 5 % CO2. The MTT medium is then removed and cells are lysed (e.g. by adding 10 % SDS solution to each well) overnight. After shaking, the absorption is measured at i.e. 600 nm with a photometer.
DATA AND REPORTING
Data evaluation
The following parameters are calculated in the KeratinoSensTM assay:
the maximal average fold induction of luciferase activity (Imax) value observed at any concentration of the tested chemical and positive control;
the EC1,5 value representing the concentration for which induction of luciferase activity is above the 1,5 fold threshold (i.e. 50 % enhanced luciferase activity) was obtained; and
the IC50 and IC30 concentration values for 50 % and 30 % reduction of cellular viability.
Fold luciferase activity induction is calculated by Equation 1, and the overall maximal fold induction (Imax) is calculated as the average of the individual repetitions.
Equation 1:

where
Lsample
is the luminescence reading in the test chemical well
Lblank
is the luminescence reading in the blank well containing no cells and no treatment
Lsolvent
is the average luminescence reading in the wells containing cells and solvent (negative) control
EC1,5 is calculated by linear interpolation according to Equation 2, and the overall EC1,5 is calculated as the geometric mean of the individual repetitions.
Equation 2:
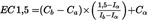
where
Ca
is the lowest concentration in μM with > 1,5 fold induction
Cb
is the highest concentration in μM with < 1,5 fold induction
Ia
is the fold induction measured at the lowest concentration with > 1,5 fold induction (mean of three replicate wells)
Ib
is the fold induction at the highest concentration with < 1,5 fold induction (mean of three replicate wells)
Viability is calculated by Equation 3:
Equation 3:

where
Vsample
is the MTT-absorbance reading in the test chemical well
Vblank
is the MTT-absorbance reading in the blank well containing no cells and no treatment
Vsolvent
is the average MTT-absorbance reading in the wells containing cells and solvent (negative) control
IC50 and IC30 are calculated by linear interpolation according to Equation 4, and the overall IC50 and IC30 are calculated as the geometric mean of the individual repetitions.
Equation 4:

where
X
is the % reduction at the concentration to be calculated (50 and 30 for IC50 and IC30)
Ca
is the lowest concentration in μM with > x % reduction in viability
Cb
is the highest concentration in μM with < x % reduction in viability
Va
is the % viability at the lowest concentration with > x % reduction in viability
Vb
is the % viability at the highest concentration with < x % reduction in viability
For each concentration showing > 1,5 fold luciferase activity induction, statistical significance is calculated (e.g. by a two-tailed Student's t-test), comparing the luminescence values for the three replicate samples with the luminescence values in the solvent (negative) control wells to determine whether the luciferase activity induction is statistically significant (p < 0,05). The lowest concentration with > 1,5 fold luciferase activity induction is the value determining the EC1,5 value. It is checked in each case whether this value is below the IC30 value, indicating that there is less than 30 % reduction in cellular viability at the EC1,5 determining concentration.
It is recommended that data are visually checked with the help of graphs. If no clear dose-response curve is observed, or if the dose-response curve obtained is biphasic (i.e. crossing the threshold of 1,5 twice), the experiment should be repeated to verify whether this is specific to the test chemical or due to an experimental artefact. In case the biphasic response is reproducible in an independent experiment, the lower EC1,5 value (the concentration when the threshold of 1,5 is crossed the first time) should be reported.
In the rare cases where a statistically non-significant induction above 1,5 fold is observed followed by a higher concentration with a statistically significant induction, results from this repetition are only considered as valid and positive if the statistically significant induction above the threshold of 1,5 was obtained for a non-cytotoxic concentration.
Finally, for test chemicals generating a 1,5 fold or higher induction already at the lowest test concentration of 0,98 μM, the EC1,5 value of < 0,98 is set based on visual inspection of the dose-response curve.
Acceptance criteria
The following acceptance criteria should be met when using the KeratinoSensTM assay. First, the luciferase activity induction obtained with the positive control, cinnamic aldehyde, should be statistically significant above the threshold of 1,5 (e.g. using a T-test) in at least one of the tested concentrations (from 4 to 64 μM).
Second, the EC1,5 value should be within two standard deviations of the historical mean of the testing facility (e.g. between 7 μM and 30 μM based on the validation dataset) which should be regularly updated. In addition, the average induction in the three replicates for cinnamic aldehyde at 64 μM should be between 2 and 8. If the latter criterion is not fulfilled, the dose-response of cinnamic aldehyde should be carefully checked, and tests may be accepted only if there is a clear dose-response with increasing luciferase activity induction at increasing concentrations for the positive control.
Finally, the average coefficient of variation of the luminescence reading for the negative (solvent) control DMSO should be below 20 % in each repetition which consists of 6 wells tested in triplicate. If the variability is higher, results should be discarded.
Interpretation of results and prediction model
A KeratinoSensTM prediction is considered positive if the following 4 conditions are all met in 2 of 2 or in the same 2 of 3 repetitions, otherwise the KeratinoSensTM prediction is considered negative (Figure 1):
1.
the Imax is higher than (>) 1,5 fold and statistically significantly different as compared to the solvent (negative) control (as determined by a two-tailed, unpaired Student's t-test);
2.
the cellular viability is higher than (>) 70 % at the lowest concentration with induction of luciferase activity above 1,5 fold (i.e. at the EC1,5 determining concentration);
3.
the EC1,5 value is less than (<) 1 000 μM (or < 200 μg/ml for test chemicals with no defined MW);
4.
there is an apparent overall dose-response for luciferase induction (or a biphasic response as mentioned under paragraph 33).
If in a given repetition, all of the three first conditions are met but a clear dose-response for the luciferase induction cannot be observed, then the result of that repetition should be considered inconclusive and further testing may be required (Figure 1). In addition, a negative result obtained with concentrations < 1 000 μM (or < 200 μg/ml for test chemicals with no defined MW) should also be considered as inconclusive (see paragraph 11).
Figure 1 Prediction model used in the KeratinoSensTM assay. A KeratinoSensTM prediction should be considered in the framework of an IATA and in accordance with the provision of paragraphs 9 and 11

In rare cases, test chemicals which induce the luciferase activity very close to the cytotoxic levels can be positive in some repetitions at non-cytotoxic levels (i.e. EC1,5 determining concentration below (<) the IC30), and in other repetitions only at cytotoxic levels (i.e. EC1,5 determining concentration above (>) the IC30). Such test chemicals shall be retested with more narrow dose-response analysis using a lower dilution factor (e.g. 1,33 or √2 (= 1,41) fold dilution between wells), to determine if induction has occurred at cytotoxic levels or not (9).
Test report
The test report should include the following information:
Test chemical
Mono-constituent substance
Chemical identification, such as IUPAC or CAS name(s), CAS number(s), SMILES or InChI code, structural formula, and/or other identifiers;
Physical appearance, water solubility, DMSO solubility, molecular weight, and additional relevant physicochemical properties, to the extent available;
Purity, chemical identity of impurities as appropriate and practically feasible, etc;
Treatment prior to testing, if applicable (e.g. warming, grinding);
Concentration(s) tested;
Storage conditions and stability to the extent available.
Multi-constituent substance, UVCB and mixture:
Characterisation as far as possible by e.g. chemical identity (see above), purity, quantitative occurrence and relevant physicochemical properties (see above) of the constituents, to the extent available;
Physical appearance, water solubility, DMSO solubility and additional relevant physicochemical properties, to the extent available;
Molecular weight or apparent molecular weight in case of mixtures/polymers of known compositions or other information relevant for the conduct of the study;
Treatment prior to testing, if applicable (e.g. warming, grinding);
Concentration(s) tested;
Storage conditions and stability to the extent available.
Controls
Positive control
Chemical identification, such as IUPAC or CAS name(s), CAS number(s), SMILES or InChI code, structural formula, and/or other identifiers;
Physical appearance, water solubility, DMSO solubility, molecular weight, and additional relevant physicochemical properties, to the extent available and where applicable;
Purity, chemical identity of impurities as appropriate and practically feasible, etc;
Treatment prior to testing, if applicable (e.g. warming, grinding);
Concentration(s) tested;
Storage conditions and stability to the extent available;
Reference to historical positive control results demonstrating suitable run acceptance criteria, if applicable.
Negative (vehicle) control
Chemical identification, such as IUPAC or CAS name(s), CAS number(s), and/or other identifiers;
Purity, chemical identity of impurities as appropriate and practically feasible, etc;
Physical appearance, molecular weight, and additional relevant physicochemical properties in the case other negative controls / vehicles than those mentioned in this test method are used and to the extent available;
Storage conditions and stability to the extent available;
Justification for choice of solvent for each test chemical.
Test method conditions
Name and address of the sponsor, test facility and study director;
Description of test method used;
Cell line used, its storage conditions and source (e.g. the facility from which they were obtained);
Passage number and level of confluence of cells used for testing;
Cell counting method used for seeding prior to testing and measures taken to ensure homogeneous cell number distribution (cf. paragraph 20);
Luminometer used (e.g. model), including instrument settings, luciferase substrate used, and demonstration of appropriate luminescence measurements based on the control test described in Appendix 3;
The procedure used to demonstrate proficiency of the laboratory in performing the test method (e.g. by testing of proficiency substances) or to demonstrate reproducible performance of the test method over time.
Test procedure
Number of repetitions and replicates used;
Test chemical concentrations, application procedure and exposure time used (if different than the one recommended)
Description of evaluation and decision criteria used;
Description of study acceptance criteria used;
Description of any modifications of the test procedure.
Results
Tabulation of Imax, EC1,5 and viability values (i.e. IC50, IC30) obtained for the test chemical and for the positive control for each repetition as well as the mean values (Imax: average; EC1,5 and viability values: geometric mean) and SD calculated using data from all individual repetitions and an indication of the rating of the test chemical according to the prediction model;
Coefficient of variation obtained with the luminescence readings for the negative control for each experiment;
A graph depicting dose-response curves for induction of luciferase activity and viability;
Description of any other relevant observations, if applicable.
Discussion of the results
Discussion of the results obtained with the KeratinoSensTM assay;
Consideration of the test method results within the context of an IATA, if other relevant information is available.
Conclusion
LITERATURE:
(1)
United Nations (UN) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS), Fifth revised edition, UN New York and Geneva, 2013. Available at: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
(2)
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. OECD Environment, Health and Safety publications, Series on Testing and Assessment No. 168. OECD, Paris.
(3)
Adler S., Basketter D., Creton S., Pelkonen O., van Benthem J., Zuang V., Andersen K.E., Angers-Loustau A., Aptula A., Bal-Price A., Benfenati E., Bernauer U., Bessems J., Bois F.Y., Boobis A., Brandon E., Bremer S., Broschard T., Casati S., Coecke S., Corvi R., Cronin M., Daston G., Dekant W., Felter S., Grignard E., Gundert-Remy U., Heinonen T., Kimber I., Kleinjans J., Komulainen H., Kreiling R., Kreysa J., Leite S.B., Loizou G., Maxwell G., Mazzatorta P., Munn S., Pfuhler S., Phrakonkham P., Piersma A., Poth A., Prieto P., Repetto G., Rogiers V., Schoeters G., Schwarz M., Serafimova R., Tähti H., Testai E., van Delft J., van Loveren H., Vinken M., Worth A., Zaldivar J.M. (2011). Alternative (non-animal) methods for cosmetics testing: current status and future prospects-2010. Archives of Toxicology 85, 367-485.
(4)
Chapter B.42 of this Annex: Skin sensitization: Local Lymph Node assay.
(5)
Chapter B.6 of this Annex: Skin Sensitisation.
(6)
Chapter B.50 of this Annex: Skin sensitization: Local Lymph Node assay: DA.
(7)
Chapter B.51 of this Annex: Skin sensitization: Local Lymph Node assay: BrdU-ELISA.
(8)
Natsch A. (2010). The Nrf2-Keap1-ARE Toxicity Pathway as a Cellular Sensor for Skin Sensitizers-Functional Relevance and Hypothesis on Innate Reactions to Skin Sensitizers. Toxicological Sciences 113, 284-292.
(9)
Emter R., Ellis G., Natsch A. (2010). Performance of a novel keratinocyte-based reporter cell line to screen skin sensitizers in vitro. Toxicology and Applied Pharmacology 245, 281-290.
(10)
Dinkova-Kostova A.T., Holtzclaw W.D., Kensler T.W. (2005). The role of Keap1 in cellular protective responses. Chem. Res. Toxicol. 18, 1779-1791.
(11)
Kansanen E., Kuosmanen S.M., Leinonen H., Levonen A.L. (2013). The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 1(1), 45-49.
(12)
Natsch A., Bauch C., Foertsch L., Gerberick F., Normann K., Hilberer A., Inglis H., Landsiedel R., Onken S., Reuter H., Schepky A., Emter R. (2011). The intra- and inter-laboratory reproducibility and predictivity of the KeratinoSens assay to predict skin sensitizers in vitro: results of a ring-study in five laboratories. Toxicol. In Vitro 25, 733-744.
(13)
Natsch A., Ryan C.A., Foertsch L., Emter R., Jaworska J., Gerberick G.F., Kern P. (2013). A dataset on 145 chemicals tested in alternative assays for skin sensitization undergoing prevalidation. Journal of Applied Toxicology, 33, 1337-1352.
(14)
EURL-ECVAM (2014). Recommendation on the KeratinoSensTM assay for skin sensitisation testing, 42 pp. Available at: http://ihcp.jrc.ec.europa.eu/our_labs/eurl-ecvam/eurl-ecvam-recommendations/recommendation-keratinosens-skin-sensitisation.
(15)
DB-ALM (INVITTOX) (2013) Protocol 155: KeratinoSensTM., 17 pp. Available: http://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index
(16)
Natsch A., Emter R., Ellis G. (2009). Filling the concept with data: integrating data from different in vitro and in silico assays on skin sensitizers to explore the battery approach for animal-free skin sensitization testing. Toxicol. Sci. 107, 106-121.
(17)
Ball N., Cagen S., Carrillo J.C., Certa H., Eigler D., Emter R., Faulhammer F., Garcia C., Graham C., Haux C., Kolle S.N., Kreiling R., Natsch A., Mehling A. (2011). Evaluating the sensitization potential of surfactants: integrating data from the local lymph node assay, guinea pig maximization test, and in vitro methods in a weight-of-evidence approach. Regul. Toxicol. Pharmacol. 60, 389-400.
(18)
Bauch C., Kolle S.N., Ramirez T., Eltze T., Fabian E., Mehling A., Teubner W., van Ravenzwaay B., Landsiedel R. (2012). Putting the parts together: combining in vitro methods to test for skin sensitizing potentials. Regul. Toxicol. Pharmacol. 63, 489-504.
(19)
Jaworska J., Dancik Y., Kern P., Gerberick F., Natsch A. (2013). Bayesian integrated testing strategy to assess skin sensitization potency: from theory to practice. J Appl Toxicol. 33, 1353-1364.
(20)
Andres E., Sa-Rocha V.M., Barrichello C., Haupt T., Ellis G., Natsch A. (2013). The sensitivity of the KeratinoSensTM assay to evaluate plant extracts: A pilot study. Toxicology In Vitro 27, 1220-1225.
(21)
Fabian E., Vogel D., Blatz V., Ramirez T., Kolle S., Eltze T., van Ravenzwaay B., Oesch F., Landsiedel R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch. Toxicol. 87, 1683-1969.
(22)
Thorne N., Inglese J., Auld D.S. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology. Chemistry and Biology 17, 646-657.
(23)
OECD (2012). BG1Luc Estrogen Receptor Transactivation Test Method for Identifying Estrogen Receptor Agonists and Antagonists. OECD Guidelines for Chemical Testing No. 457. OECD, Paris.
(24)
ECETOC (2003). Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals (Technical Report No. 87).
(25)
Gildea L.A., Ryan C.A., Foertsch L.M., Kennedy J.M., Dearman R.J., Kimber I., Gerberick G.F. (2006). Identification of gene expression changes induced by chemical allergens in dendritic cells: opportunities for skin sensitization testing. J. Invest. Dermatol., 126, 1813-1822.
(26)
Ryan C.A., Gildea L.A., Hulette B.C., Dearman R.J., Kimber I., Gerberick G.F. (2004). Gene expressing changes in peripheral blood-derived dendritic cells following exposure to a contact allergen. Toxicol. Lett. 150, 301-316.
(27)
Emter R., van der Veen J.W., Adamson G., Ezendam J., van Loveren H., Natsch A. (2013). Gene expression changes induced by skin sensitizers in the KeratinoSens™ cell line: Discriminating Nrf2-dependent and Nrf2-independent events. Toxicol. in vitro 27, 2225-2232.
(28)
OECD (2015). Performance Standards for assessment of proposed similar or modified in vitro skin sensitisation ARE-Nrf2 luciferase test methods. OECD Environment, Health and Safety publications, Series on Testing and Assessment N0 213, OECD, Paris.
(29)
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Environment, Health and Safety publications, Series on Testing and Assessment No.34. OECD, Paris, France.
(30)
NAFTA (North American Free Trade Agreement) (2012). Technical Working Group on Pesticides — (Quantitative) Structure Activity Relationship ((Q)SAR) Guidance Document. 186 pp. http://www.epa.gov/oppfead1/international/naftatwg/guidance/qsar-guidance.pdf
(1)
Regulation (EC) No 1272/2008 of the European Parliament and of the Council of 16 December 2008 on classification, labelling and packaging of substances and mixtures, amending and repealing Directives 67/548/EEC and 1999/45/EC, and amending Regulation (EC) No 1907/2006 (OJ L 353, 31.12.2008, p. 1).
Options/Help
Print Options
PrintThe Whole Regulation
PrintThe Whole Annex
PrintThis Division only
You have chosen to open the Whole Regulation
The Whole Regulation you have selected contains over 200 provisions and might take some time to download. You may also experience some issues with your browser, such as an alert box that a script is taking a long time to run.
Would you like to continue?
You have chosen to open Schedules only
The Schedules you have selected contains over 200 provisions and might take some time to download. You may also experience some issues with your browser, such as an alert box that a script is taking a long time to run.
Would you like to continue?
Legislation is available in different versions:
Latest Available (revised):The latest available updated version of the legislation incorporating changes made by subsequent legislation and applied by our editorial team. Changes we have not yet applied to the text, can be found in the ‘Changes to Legislation’ area.
Original (As adopted by EU): The original version of the legislation as it stood when it was first adopted in the EU. No changes have been applied to the text.
Opening Options
Different options to open legislation in order to view more content on screen at once
More Resources
Access essential accompanying documents and information for this legislation item from this tab. Dependent on the legislation item being viewed this may include:
- the original print PDF of the as adopted version that was used for the EU Official Journal
- lists of changes made by and/or affecting this legislation item
- all formats of all associated documents
- correction slips
- links to related legislation and further information resources
More Resources
Use this menu to access essential accompanying documents and information for this legislation item. Dependent on the legislation item being viewed this may include:
- the original print PDF of the as adopted version that was used for the print copy
- correction slips
Click 'View More' or select 'More Resources' tab for additional information including:
- lists of changes made by and/or affecting this legislation item
- confers power and blanket amendment details
- all formats of all associated documents
- links to related legislation and further information resources
 All content is available under the Open Government Licence v3.0 except where otherwise stated. This site additionally contains content derived from EUR-Lex, reused under the terms of the Commission Decision 2011/833/EU on the reuse of documents from the EU institutions. For more information see the EUR-Lex public statement on re-use.
All content is available under the Open Government Licence v3.0 except where otherwise stated. This site additionally contains content derived from EUR-Lex, reused under the terms of the Commission Decision 2011/833/EU on the reuse of documents from the EU institutions. For more information see the EUR-Lex public statement on re-use.