ANNEX VIIIU.K. Methods for the quantitative analysis of binary and ternary textile fibre mixtures (referred to in Article 19(1))
CHAPTER 3U.K.
QUANTITATIVE ANALYSIS OF TERNARY TEXTILE FIBRE MIXTURES U.K.
INTRODUCTIONU.K.
In general, the methods of quantitative chemical analysis are based on the selective solution of the individual components. There are four possible variants of this method:
Using two different test specimens, a component (a) is dissolved from the first test specimen, and another component (b) from the second test specimen. The insoluble residues of each specimen are weighed and the percentage of each of the two soluble components is calculated from the respective losses in mass. The percentage of the third component (c) is calculated by difference.
Using two different test specimens, a component (a) is dissolved from the first test specimen and two components (a and b) from the second test specimen. The insoluble residue of the first test specimen is weighed and the percentage of the component (a) is calculated from the loss in mass. The insoluble residue of the second test specimen is weighed; it corresponds to component (c). The percentage of the third component (b) is calculated by difference.
Using two different test specimens, two components (a and b) are dissolved from the first test specimen and two components (b and c) from the second test specimen. The insoluble residues correspond to the two components (c) and (a) respectively. The percentage of the third component (b) is calculated by difference.
Using only one test specimen, after removal of one of the components, the insoluble residue formed by the two other fibres is weighed and the percentage of the soluble component is calculated from the loss in mass. One of the two fibres of the residue is dissolved, the insoluble component is weighed and the percentage of the second soluble component is calculated from the loss in mass.
Where a choice is possible, it is advisable to use one of the first three variants.
Where chemical analysis is used, the expert responsible for the analysis must take care to select methods employing solvents which dissolve only the correct fibre(s), leaving the other fibre(s) intact.
By way of example, a table is given in Section V which contains a certain number of ternary fibre mixtures, together with methods for analysing binary fibre mixtures which can, in principle, be used for analysing these ternary fibre mixtures.
In order to reduce the possibility of error to a minimum, it is recommended that, whenever possible, chemical analysis using at least two of the four abovementioned variants shall be made.
Before proceeding with any analysis, all the fibres present in the mixture must be identified. In some chemical methods, the insoluble component of a mixture may be partially dissolved in the reagent used to dissolve the soluble component(s). Wherever possible, reagents have been chosen that have little or no effect on the insoluble fibres. If a loss in mass is known to occur during the analysis, the result shall be corrected; correction factors are given for this purpose. These factors have been determined in several laboratories by treating, with the appropriate reagent as specified in the method of analysis, fibres cleaned by the pre-treatment. These correction factors apply only to undergraded fibres and different correction factors may be necessary if the fibres have been degraded before or during processing. If the fourth variant, in which a textile fibre is subjected to the successive action of two different solvents, must be used, correction factors must be applied for possible losses in mass undergone by the fibre in the two treatments. At least two determinations shall be made, both in the case of manual separation and in the case of chemical separation.
I. General information on methods for the quantitative chemical analysis of ternary fibre mixtures U.K.
Information common to the methods given for the quantitative chemical analysis of ternary fibre mixtures.
I.1.FIELD OF APPLICATIONU.K.
The field of application of each method for analysing binary fibre mixtures specifies to which fibres the method is applicable (see Chapter 2 relating to methods for quantitative analysis of certain binary textile fibre mixtures).
I.2.PRINCIPLEU.K.
After the identification of the components of a mixture, the non-fibrous material is removed by suitable pre-treatment and then one or more of the four variants of the process of selective solution described in the introduction is applied. Except where this presents technical difficulties, it is preferable to dissolve the major fibre component so as to obtain the minor fibre component as final residue.
I.3.MATERIALS AND EQUIPMENTU.K.
I.3.1.ApparatusU.K.
I.3.1.1.Filter crucibles and weighing bottles large enough to contain such crucibles, or any other apparatus giving identical results.U.K.
I.3.1.2.Vacuum flask.U.K.
I.3.1.3.Desiccator containing self-indicating silica gel.U.K.
I.3.1.4.Ventilated oven for drying specimens at 105 ± 3 °C.U.K.
I.3.1.5.Analytical balance, accurate to 0,0002 g.U.K.
I.3.1.6.Soxhlet extractor or other apparatus giving identical results.U.K.
I.3.2.ReagentsU.K.
I.3.2.1.Light petroleum, redistilled, boiling range 40 to 60 °C.U.K.
I.3.2.2.Other reagents are specified in the appropriate sections of each method.U.K.
I.3.2.3.Distilled or deionised water.U.K.
I.3.2.4.Acetone.U.K.
I.3.2.5.Orthophosphoric acid.U.K.
I.3.2.6.Urea.U.K.
I.3.2.7.Sodium bicarbonate.U.K.
All reagents used shall be chemically pure.
I.4.CONDITIONING AND TESTING ATMOSPHEREU.K.
Because dry masses are determined, it is unnecessary to condition the specimen or to conduct analyses in a conditioned atmosphere.
I.5.LABORATORY TEST SAMPLEU.K.
Take a laboratory test sample that is representative of the laboratory bulk sample and sufficient to provide all the specimens, each of at least 1 g, that are required.
I.6.PRE-TREATMENT OF LABORATORY TEST SAMPLE(1) U.K.
Where a substance not to be taken into account in the percentage calculations (see Article 19) is present, it shall first be removed by a suitable method that does not affect any of the fibre constituents.
For this purpose, non-fibrous matter which can be extracted with light petroleum and water is removed by treating the laboratory test sample in a Soxhlet extractor with light petroleum for 1 hour at a minimum rate of six cycles per hour. Allow the light petroleum to evaporate from the laboratory test sample, which is then extracted by direct treatment consisting in soaking the laboratory test sample in water at room temperature for 1 hour and then soaking it in water at 65 ± 5 °C for a further hour, agitating the liquor from time to time. Use a liquor: laboratory test sample ratio of 100:1. Remove the excess water from the laboratory test sample by squeezing, suction or centrifuging and then allow the laboratory test sample to become air-dry.
In the case of elastolefin or fibre mixtures containing elastolefin and other fibres (wool, animal hair, silk, cotton, flax (or linen), true hemp, jute, abaca, alfa, coir, broom, ramie, sisal, cupro, modal, protein, viscose, acrylic, polyamide or nylon, polyester, elastomultiester) the procedure just described shall be slightly modified, in fact light petroleum ether shall be replaced by acetone.
Where non-fibrous matter cannot be extracted with light petroleum and water, it shall be removed by substituting for the water method described above a suitable method that does not substantially alter any of the fibre constituents. However, for some unbleached, natural vegetable fibres (e.g. jute, coir) it is to be noted that normal pre-treatment with light petroleum and water does not remove all the natural non-fibrous substances; nevertheless additional pre-treatment is not applied unless the sample contains finishes insoluble in both light petroleum and water.
Analysis reports shall include full details of the methods of pre-treatment used.
I.7.TEST PROCEDUREU.K.
I.7.1.General instructionsU.K.
I.7.1.1.DryingU.K.
Conduct all drying operations for not less than 4 hours and not more than 16 hours at 105 ± 3 °C in a ventilated oven with the oven door closed throughout. If the drying period is less than 14 hours, the specimen must be checkweighed to determine whether its mass is constant. The mass may be considered as constant if, after a further drying period of 60 minutes, its variation is less than 0,05 %.
Avoid handling crucibles and weighing bottles, specimens or residues with bare hands during the drying, cooling and weighing operations.
Dry specimens in a weighing bottle with its cover beside it. After drying, stopper the weighing bottle before removing it from the oven, and transfer it quickly to the desiccator.
Dry the filter crucible in a weighing bottle with its cover beside it in the oven. After drying, close the weighing bottle and transfer it quickly to the desiccator.
Where apparatus other than a filter crucible is used, drying operations shall be conducted in the oven so as to determine the dry mass of the fibres without loss.
I.7.1.2.CoolingU.K.
Conduct all cooling operations in the desiccator, placed beside the balance, until the cooling of the weighing bottles is complete, and in any case for not less than 2 hours.
I.7.1.3.WeighingU.K.
After cooling, complete the weighing of the weighing bottle within 2 minutes of its removal from the desiccator; weigh to an accuracy of 0,0002 g.
I.7.2.ProcedureU.K.
Take from the pre-treated laboratory test sample a test specimen of at least 1 g (in mass). Cut yarn or cloth into lengths of about 10 mm, dissected as much as possible. Dry the specimen in a weighing bottle, cool it in the desiccator and weigh it. Transfer the specimen to the glass vessel specified in the appropriate section of the Union method, reweigh the weighing bottle immediately and obtain the dry mass of the specimen by difference; complete the test as specified in the appropriate section of the applicable method. Examine the residue microscopically to check that the treatment has in fact completely removed the soluble fibre(s).
I.8.CALCULATION AND EXPRESSION OF RESULTSU.K.
Express the mass of each component as a percentage of the total mass of fibre in the mixture. Calculate the results on the basis of dean dry mass, adjusted by (a) the agreed allowances and (b) the correction factors necessary to take account of loss of non-fibrous matter during pre-treatment and analysis.
I.8.1.Calculation of percentages of mass of clean dry fibres disregarding loss of fibre mass during pre-treatment.U.K.
I.8.1.1.- VARIANT 1 -U.K.
Formulae to be applied where a component of the mixture is removed from one specimen and another component from a second specimen:


P 3% = 100 – (P 1% + P 2%)
is the percentage of the first clean dry component (component in the first specimen dissolved in the first reagent),
is the percentage of the second clean dry component (component in the second specimen dissolved in the second reagent),
is the percentage of the third clean dry component (component undissolved in both specimens),
is the dry mass of the first specimen after pre-treatment,
is the dry mass of the second specimen after pre-treatment,
is the dry mass of the residue after removal of the first component from the first specimen in the first reagent,
is the dry mass of the residue after removal of the second component from the second specimen in the second reagent,
is the correction factor for loss in mass in the first reagent, of the second component undissolved in the first specimen(2);
is the correction factor for loss in mass in the first reagent, of the third component undissolved in the first specimen,
is the correction factor for loss in mass in the second reagent, of the first component undissolved in the second specimen,
is the correction factor for loss in mass in the second reagent, of the third component undissolved in the second specimen.
I.8.1.2.- VARIANT 2 -U.K.
Formulae to be applied where a component (a) is removed from the first test specimen, leaving as residue the other two components (b + c), and two components (a + b) are removed from the second test specimen, leaving as residue the third component (c):
P 1% = 100 – (P 2% + P 3%)


is the percentage of the first clean dry component (component in the first specimen dissolved in the first reagent),
is the percentage of the second clean dry component (component soluble, at the same time as the first component of the second specimen, in the second reagent),
is the percentage of the third clean dry component (component undissolved in both specimens),
is the dry mass of the first specimen after pre-treatment,
is the dry mass of the second specimen after pre-treatment,
is the dry mass of the residue after removal of the first component from the first specimen in the first reagent,
is the dry mass of the residue after removal of the first and second components from the second specimen in the second reagent,
is the correction factor for loss in mass in the first reagent, of the second component undissolved in the first specimen,
is the correction factor for loss in mass in the first reagent, of the third component undissolved in the first specimen,
is the correction factor for loss in mass in the second reagent, of the third component undissolved in the second specimen.
I.8.1.3.- VARIANT 3 -U.K.
Formulae to be applied where two components (a + b) are removed from a specimen, leaving as residue the third component (c), then two components (b + c) are removed from another specimen, leaving as residue the first component (a):

P 2% = 100 – (P 1% + P 3%)

is the percentage of the first clean dry component (component dissolved by the reagent),
is the percentage of the second clean dry component (component dissolved by the reagent),
is the percentage of the third clean dry component (component dissolved in the second specimen by the reagent),
is the dry mass of the first specimen after pre-treatment,
is the dry mass of the second specimen after pre-treatment,
is the dry mass of the residue after the removal of the first and second components from the first specimen with the first reagent,
is the dry mass of the residue after the removal of the second and third components from the second specimen with the second reagent,
is the correction factor for loss in mass in the first reagent of the third component undissolved in the first specimen,
is the correction factor for loss in mass in the second reagent of the first component undissolved in the second specimen.
I.8.1.4.- VARIANT 4 -U.K.
Formulae to be applied where two components are successively removed from the mixture using the same specimen:
P 1% = 100 – (P 2% + P 3%)

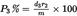
is the percentage of the first clean dry component (first soluble component),
is the percentage of the second clean dry component (second soluble component),
is the percentage of the third clean dry component (insoluble component),
is the dry mass of the specimen after pre-treatment,
is the dry mass of the residue after elimination of the first component by the first reagent,
is the dry mass of the residue after elimination of the first and second component by the first and second reagents,
is the correction factor for loss in mass of the second component in the first reagent,
is the correction factor for loss in mass of the third component in the first reagent,
is the correction factor for loss in mass of the third component in the first and second reagents(3).
I.8.2.Calculation of the percentage of each component with adjustment by agreed allowances and, where appropriate, correction factors for losses in mass during pre-treatment operations:U.K.
Given:



then:



is the percentage of the first clean dry component, including moisture content and loss in mass during pre-treatment,
is the percentage of the second clean dry component, including moisture content and loss in mass during pre-treatment,
is the percentage of the third clean dry component, including moisture content and loss in mass during pre-treatment,
is the percentage of the first clean dry component obtained by one of the formulae given in I.8.1,
is the percentage of the second clean dry component obtained by one of the formulae given in I.8.1,
is the percentage of the third clean dry component obtained by one of the formulae given in I.8.1,
is the agreed allowance of the first component,
is the agreed allowance of the second component,
is the agreed allowance of the third component,
is the percentage of loss in mass of the first component during pre-treatment,
is the percentage of loss in mass of the second component during pre-treatment,
is the percentage of loss in mass of the third component during pre-treatment.
Where a special pre-treatment is used the values b1, b2 and b3 shall be determined, if possible, by submitting each of the pure fibre constituents to the pre-treatment applied in the analysis. Pure fibres are those free from all non-fibrous material except those which they normally contain (either naturally or because of the manufacturing process), in the state (unbleached, bleached) in which they are found in the material to be analysed.
Where no clean separate constituent fibres used in the manufacture of the material to be analysed are available, average values of b1, b2 and b3 as obtained from tests performed on clean fibres similar to those in the mixture under examination, must be used.
If normal pre-treatment by extraction with light petroleum and water is applied, correction factors b1, b2 and b3 may generally be ignored, except in the case of unbleached cotton, unbleached flax (or linen) and unbleached hemp where the loss due to pre-treatment is usually accepted as 4 % and in the case of polypropylene as 1 %.
In the case of other fibres, losses due to pre-treatment are usually disregarded in calculations.
I.8.3.Note:U.K.
Calculation examples are given in Section IV.
II. Method of quantitative analysis by manual separation of ternary fibre mixtures U.K.
II.1.FIELD OF APPLICATIONU.K.
This method is applicable to textile fibres of all types provided they do not form an intimate mixture and that it is possible to separate them by hand.
II.2.PRINCIPLEU.K.
After identification of the textile components, the non-fibrous matter is removed by a suitable pre-treatment and then the fibres are separated by hand, dried and weighed in order to calculate the proportion of each fibre in the mixture.
II.3.APPARATUSU.K.
II.3.1.Weighing bottles or other apparatus giving identical results.U.K.
II.3.2.Desiccator containing self-indicating silica gel.U.K.
II.3.3.Ventilated oven for drying specimens at 105 ± 3 °C.U.K.
II.3.4.Analytical balance accurate to 0,0002 g.U.K.
II.3.5.Soxhlet extractor, or other apparatus giving identical results.U.K.
II.3.6.Needle.U.K.
II.3.7.Twist tester or similar apparatus.U.K.
II.4.REAGENTSU.K.
II.4.1.Light petroleum, redistilled, boiling range 40 to 60 °C.U.K.
II.4.2.Distilled or deionised water.U.K.
II.5.CONDITIONING AND TESTING ATMOSPHEREU.K.
See I.4.
II.6.LABORATORY TEST SAMPLEU.K.
See I.5.
II.7.PRE-TREATMENT OF LABORATORY TEST SAMPLESU.K.
See I.6.
II.8.PROCEDUREU.K.
II.8.1.Analysis of yarnU.K.
Take from the pre-treated laboratory test sample a specimen of mass not less than 1 g. For a very fine yarn, the analysis may be made on a minimum length of 30 m, whatever its mass.
Cut the yarn into pieces of a suitable length and separate the fibre types by means of a needle and, if necessary, a twist tester. The fibre types so obtained are placed in pre-weighed weighing bottles and dried at 105 ± 3 °C to constant mass, as described in I.7.1 and I.7.2.
II.8.2.Analysis of clothU.K.
Take from the pre-treated laboratory test sample a specimen of mass not less than 1 g, not including a selvedge with edges carefully trimmed to avoid fraying and running parallel with weft or warp yarns, or in the case of knitted fabrics in the line of the wales and courses. Separate the different types of fibres and collect them in pre-weighed weighing bottles and proceed as described in II.8.1.
II.9.CALCULATION AND EXPRESSION OF RESULTSU.K.
Express the mass of each component fibre as a percentage of the total mass of the fibres in the mixture. Calculate the results on the basis of clean dry mass, adjusted by (a) the agreed allowances and (b) the correction factors necessary to take account of losses in mass during pre-treatment operations.
II.9.1.Calculation of percentage masses of clean dry fibre, disregarding loss in fibre mass during pre-treatment:U.K.


P 3% = 100 – (P 1% + P 2%)
is the percentage of the first clean dry component,
is the percentage of the second clean dry component,
is the percentage of the third clean dry component,
is the clean dry mass of the first component,
is the clean dry mass of the second component,
is the clean dry mass of the third component.
II.9.2.For calculation of the percentage of each component with adjustment by agreed allowances and, where appropriate, by correction factors for losses in mass during pre-treatment: see I.8.2.U.K.
III. Method of quantitative analysis of ternary fibre mixtures by a combination of manual separation and chemical separation U.K.
Wherever possible, manual separation shall be used, taking account of the proportions of components separated before proceeding to any chemical treatment of each of the separate components.
III.1.PRECISION OF THE METHODSU.K.
The precision indicated in each method of analysis of binary fibre mixtures relates to the reproducibility (see Chapter 2 relating to methods for quantitative analysis of certain binary textile fibre mixtures).
Reproducibility refers to the reliability, i.e. the closeness of agreement between experimental values obtained by operators in different laboratories or at different times using the same method and obtaining individual results on specimens of an identical homogeneous mixture.
Reproducibility is expressed by confidence limits of the results for a confidence level of 95 %.
By this is meant that the difference between two results in a series of analyses made in different laboratories would, given a normal and correct application of the method to an identical and homogeneous mixture, exceed the confidence limit only in five cases out of 100.
To determine the precision of the analysis of a ternary fibre mixture the values indicated in the methods for the analysis of binary fibre mixtures which have been used to analyse the ternary fibre mixture are applied in the usual way.
Given that in the four variants of the quantitative chemical analysis of ternary fibre mixtures, provision is made for two dissolutions (using two separate specimens for the first three variants and a single specimen for the fourth variant) and, assuming that E1 and E2 denote the precision of the two methods for analysing binary fibre mixtures, the precision of the results for each component is shown in the following table:
| Component fibre | Variants | ||
|---|---|---|---|
| 1 | 2 and 3 | 4 | |
| a | E1 | E1 | E1 |
| b | E2 | E1 + E2 | E1 + E2 |
| c | E1 + E2 | E2 | E1 + E2 |
If the fourth variant is used, the degree of precision may be found to be lower than that calculated by the method indicated above, owing to possible action of the first reagent on the residue consisting of components b and c, which would be difficult to evaluate.
III.2.TEST REPORTU.K.
III.2.1.Indicate the variant(s) used to carry out the analysis, the methods, reagents and correction factors.U.K.
III.2.2.Give details of any special pre-treatments (see I.6).U.K.
III.2.3.Give the individual results and the arithmetic mean, each to the first decimal place.U.K.
III.2.4.Wherever possible, state the precision of the method for each component, calculated according to the table in Section III.1.U.K.
IV. Examples of the calculation of percentages of the components of certain ternary fibre mixtures using some of the variants described in point I.8.1. U.K.
Consider the case of a fibre mixture which gave the following components when qualitatively analysed for raw material composition: 1. carded wool; 2. nylon (polyamide); 3. unbleached cotton.
VARIANT No 1U.K.
Using this variant, that is using two different specimens and removing one component (a = wool) by dissolution from the first specimen and a second component (b = polyamide) from the second specimen, the following results can be obtained:
Dry mass of the first specimen after pre-treatment is (m1) = 1,6000 g
Dry mass of the residue after treatment with alkaline sodium hypochlorite (polyamide + cotton) (r1) = 1,4166 g
Dry mass of the second specimen after pre-treatment (m2) = 1,8000 g
Dry mass of the residue after treatment with formic acid (wool + cotton) (r2) = 0,9000 g
Treatment with alkaline sodium hypochlorite does not entail any loss in mass of polyamide, while unbleached cotton loses 3 %, therefore d1 = 1,00 and d2 = 1,03.
Treatment with formic acid does not entail any loss in mass for wool or unbleached cotton, therefore d3 and d4 = 1,00.
If the values obtained by chemical analysis and the correction factors are substituted in the formula under I.8.1.1, the following result is obtained:
P1% (wool) = [1,03/1,00 – 1,03 × 1,4166/1,6000 + (0,9000/1,8000) × (1 – 1,03/1,00)] ×100 = 10,30
P2% (polyamide) = [1,00/1,00 – 1,00 × 0,9000/1,8000 + (1,4166/1,6000) × (1 – 1,00/1,00)] × 100 = 50,00
P3% (cotton) = 100 – (10,30 + 50,00) = 39,70
The percentages of the various clean dry fibres in the mixture are as follows:
| wool | 10,3 % |
| polyamide | 50,0 % |
| cotton | 39,7 % |
These percentages must be corrected according to the formulae under I.8.2, in order to take account of the agreed allowances and the correction factors for any losses in mass after pre-treatment.
As indicated in Annex IX, the agreed allowances are as follows: carded wool 17,00 %, polyamide 6,25 %, cotton 8,50 %, also unbleached cotton shows a loss in mass of 4 %, after pre-treatment with light petroleum and water.
Therefore:
P1A% (wool) = 10,30 × [1 + (17,00 + 0,0)/100] / [10,30 × (1 + (17,00 + 0,0)/100) + 50,00 × (1 + (6,25 + 0,0)/100) + 39,70 × (1 + (8,50 + 4,0)/100)] × 100 = 10,97
P2A% (polyamide) = 50,0 × [(1 + (6,25 + 0,0)/100)/109,8385] × 100 = 48,37
P3A% (cotton) = 100 – (10,97 + 48,37) = 40,66
The raw material composition of the yarn is therefore as follows:
| polyamide | 48,4 % |
| cotton | 40,6 % |
| wool | 11,0 % |
| 100,0 % |
VARIANT No 4U.K.
Consider the case of a fibre mixture which when qualitatively analysed gave the following components: carded wool, viscose, unbleached cotton.
Suppose that using variant 4, that is successively removing two components from the mixture of one single specimen, the following results are obtained:
Dry mass of the specimen after pre-treatment (m) = 1,6000 g
Dry mass of the residue after treatment with alkaline sodium hypochlorite (viscose + cotton) (r1) = 1,4166 g
Dry mass of the residue after the second treatment of the residue r1 with zinc chloride/formic acid (cotton) (r2) = 0,6630 g
Treatment with alkaline sodium hypochlorite does not entail any loss in mass of viscose, while unbleached cotton loses 3 %, therefore d1 =1,00 and d2 = 1,03.
As a result of treatment with formic acid-zinc chloride, the mass of cotton increases by 4 %, so that d3 = 1,03 × 0,96 = 0,9888, rounded to 0,99, (d3 being the correction factor for the respective loss or increase in mass of the third component in the first and second reagents).
If the values obtained by chemical analysis and the correction factors are substituted in the formulae given in I.8.1.4, the following result is obtained:
P2% (viscose) = 1,00 × (1,4166/1,6000) × 100 – (1,00/1,03) × 41,02 = 48,71 %
P3% (cotton) = 0,99 × (0,6630/1,6000) × 100 = 41,02 %
P1% (wool) = 100 – (48,71 + 41,02) = 10,27 %
As has already been indicated for Variant 1, these percentages must be corrected by the formulae indicated in point I.8.2.
P1A% (wool) = 10,27 × [1 + (17,0 + 0,0)/100)]/[10,27 × (1 + (17,00 + 0,0)/100) +48,71 × (1 + (13 + 0,0)/100) + 41,02 × (1 + (8,5 + 4,0)/100)] × 100 = 10,61 %
P2A% (viscose) = 48,71 × [1 + (13 + 0,0)/100] / 113,2057 × 100 = 48,62 %
P3A% (cotton) = 100 – (10,61 + 48,62) = 40,77 %
The raw material composition of the mixture is therefore as follows:
| viscose | 48,6 % |
| cotton | 40,8 % |
| wool | 10,6 % |
| — | |
| 100,0 % |
V. Table of typical ternary fibre mixtures which may be analysed using Union methods of analysis of binary fibre mixtures (for illustration purposes) U.K.
| Mixture No | Component fibres | Variant | Number of method used and reagent for binary fibre mixtures | ||
|---|---|---|---|---|---|
| Component 1 | Component 2 | Component 3 | |||
| 1. | wool or hair | viscose, cupro or certain types of modal | cotton | 1 and/or 4 | 2. (hypochlorite) and 3. (zinc chloride/formic acid) |
| 2. | wool or hair | polyamide or nylon | cotton, viscose, cupro or modal | 1 and/or 4 | 2. (hypochlorite) and 4. (formic acid, 80 % m/m) |
| 3. | wool, hair or silk | certain other fibres | viscose, cupro modal or cotton | 1 and/or 4 | 2. (hypochlorite) and 9. (carbon disulphide/acetone 55,5/44,5 % v/v) |
| 4. | wool or hair | polyamide or nylon | polyester, polypropylene, acrylic or glass fibre | 1 and/or 4 | 2. (hypochlorite) and 4. (formic acid, 80 % m/m) |
| 5. | wool, hair or silk | certain other fibres | polyester, acrylic, polyamide or nylon or glass fibre | 1 and/or 4 | 2. (hypochlorite) and 9. (carbon disulphide/acetone 55,5/44,5 % v/v) |
| 6. | silk | wool or hair | polyester | 2 | 11. (sulphuric acid 75 % m/m) and 2. (hypochlorite) |
| 7. | polyamide or nylon | acrylic or certain other fibres | cotton, viscose, cupro or modal | 1 and/or 4 | 4. (formic acid 80 % m/m) and 8. (dimethylformamide) |
| 8. | certain chlorofibres | polyamide or nylon | cotton, viscose, cupro or modal | 1 and/or 4 | 8. (dimethylformamide) and 4. (formic acid, 80 % m/m) or 9. (carbon disulphide/acetone, 55,5/44,5 % v/v) and 4. (formic acid, 80 % m/m) |
| 9. | acrylic | polyamide or nylon | polyester | 1 and/or 4 | 8. (dimethylformamide) and 4. (formic acid, 80 % m/m) |
| 10. | acetate | polyamide or nylon or certain other fibres | viscose, cotton, cupro or modal | 4 | 1. (acetone) and 4. (formic acid, 80 % m/m) |
| 11. | certain chlorofibres | acrylic or certain other fibres | polyamide or nylon | 2 and/or 4 | 9. (carbon disulphide/acetone 55,5/44,5 % v/v) and 8. (dimethylformamide) |
| 12. | certain chlorofibres | polyamide or nylon | acrylic | 1 and/or 4 | 9. (carbon disulphide/acetone 55,5/44,5 % v/v) and 4. (formic acid, 80 %m/m) |
| 13. | polyamide or nylon | viscose, cupro, modal or cotton | polyester | 4 | 4. (formic acid, 80 % m/m) and 7. (sulphuric acid, 75 % m/m) |
| 14. | acetate | viscose, cupro, modal or cotton | polyester | 4 | 1. (acetone) and 7 (sulphuric acid, 75 % m/m) |
| 15. | acrylic | viscose, cupro, modal or cotton | polyester | 4 | 8. (dimethylformamide) and 7. (sulphuric acid, 75 % m/m) |
| 16. | acetate | wool, hair or silk | cotton, viscose, cupro, modal, polyamide or nylon, polyester, acrylic | 4 | 1. (acetone) and 2. (hypochlorite) |
| 17. | triacetate | wool, hair or silk | cotton, viscose, cupro, modal, polyamide or nylon, polyester, acrylic | 4 | 6. (dichloromethane) and 2. (hypochlorite) |
| 18. | acrylic | wool, hair or silk | polyester | 1 and/or 4 | 8. (dimethylformamide) and 2. (hypochlorite) |
| 19. | acrylic | silk | wool or hair | 4 | 8. (dimethylformamide) and 11. (sulphuric acid 75 % m/m) |
| 20. | acrylic | wool or hair silk | cotton, viscose, cupro or modal | 1 and/or 4 | 8. (dimethylformamide) and 2. (hypochlorite) |
| 21. | wool, hair or silk | cotton, viscose, modal, cupro | polyester | 4 | 2. (hypochlorite) and 7. (sulphuric acid 75 % m/m) |
| 22. | viscose, cupro or certain types of modal | cotton | polyester | 2 and/or 4 | 3. (zinc chloride/formic acid) and 7. (sulphuric acid 75 % m/m) |
| 23. | acrylic | viscose, cupro or certain types of modal | cotton | 4 | 8. (dimethylformamide) and 3 (zinc chloride/formic acid) |
| 24. | certain chlorofibres | viscose, cupro or certain types of modal | cotton | 1 and/or 4 | 9. (carbon disulphide/acetone, 55,5/44,5 % v/v) and 3. (zinc chloride/formic acid) or 8. (dimethylformamide) and 3. (zinc chloride/formic acid) |
| 25. | acetate | viscose, cupro or certain types of modal | cotton | 4 | 1. (acetone) and 3. (zinc chloride/formic acid) |
| 26. | triacetate | viscose, cupro or certain types of modal | cotton | 4 | 6. (dichloromethane) and 3. (zinc chloride/formic acid) |
| 27. | acetate | silk | wool or hair | 4 | 1. (acetone) and 11. (sulphuric acid 75 % m/m) |
| 28. | triacetate | silk | wool or hair | 4 | 6. (dichloromethane) and 11. (sulphuric acid 75 % m/m) |
| 29. | acetate | acrylic | cotton, viscose, cupro or modal | 4 | 1. (acetone) and 8. (dimethylformamide) |
| 30. | triacetate | acrylic | cotton, viscose, cupro or modal | 4 | 6. (dichloromethane) and 8. (dimethylformamide) |
| 31. | triacetate | polyamide or nylon | cotton, viscose, cupro or modal | 4 | 6. (dichloromethane) and 4. (formic acid 80 % m/m) |
| 32. | triacetate | cotton, viscose, cupro or modal | polyester | 4 | 6. (dichloromethane) and 7. (sulphuric acid 75 % m/m) |
| 33. | acetate | polyamide or nylon | polyester or acrylic | 4 | 1. (acetone) and 4. (formic acid 80 % m/m) |
| 34. | acetate | acrylic | polyester | 4 | 1. (acetone) and 8. (dimethylformamide) |
| 35. | certain chlorofibres | cotton, viscose, cupro or modal | polyester | 4 | 8. (dimethylformamide) and 7. (sulphuric acid 75 % m/m) or 9 (carbon disulphide/acetone, 55,5/44,5 % v/v) and 7. (sulphuric acid 75 % m/m) |
| 36. | cotton | polyester | elastolefin | 2 and/or 4 | 7. (sulphuric acid 75 % m/m) and 14. (concentrated sulphuric acid) |
| 37. | certain modacrylics | polyester | melamine | 2 and/or 4 | 8. (dimethylformamide) and 14. (concentrated sulphuric acid) |
See Chapter 1.1.
The values of d are indicated in Chapter 2 of this Annex relating to the various methods of analysing binary mixtures.
Wherever possible d3 should be determined in advance by experimental methods.