Regulation 6
SCHEDULE 2METHODS OF ANALYSIS
PART I
General
1.—(a) When two or more methods are prescribed in this part of this Schedule to determine a component of a fertiliser the choice of the method shall, except where otherwise indicated, be left to the agricultural analyst concerned; the method used must however be indicated in the certificate of analysis.
(b)Any reference to water in this Schedule means purified water as defined in the European Pharmacopoeia.
Reagents and Apparatus
2.—(a) All reagents used shall be analytical quality.
(b)For the determination of any form of nitrogen, water must be free of all nitrogenous compounds and carbon dioxide.
(c)Solutions for which no solvents are prescribed must be aqueous.
(d)Only special instruments or apparatus requiring special standards are mentioned in the descriptions of the methods of analysis.
Methods of analysis
3.—1. Preparation of the sample for analysis
2. Determination of ammoniacal nitrogen
3.a Determination of nitrate and ammoniacal nitrogen—Ulsch method
b Determination of nitrate and ammoniacal nitrogen—Arnd method
c Determination of nitrate and ammoniacal nitrogen—Devarda method
4.a Determination of nitrogen in calcium cyanamide—in the absence of nitrate
b Determination of nitrogen in calcium cyanamide—in the presence of nitrate
5. Determination of total nitrogen in urea
6. Determination of cyanamide nitrogen
7. Determination of biuret in urea
8.a Determination of different forms of nitrogen—in the presence of cyanamide nitrogen
b Determination of different forms of nitrogen—in the absence of cyanamide nitrogen
9.a Extraction of total phosphorus—by mineral acids
b Extraction of phosphorus—by 2% formic acid
c Extraction of phosphorus—by 2% citric acid
d Extraction of phosphorus—by neutral ammonium citrate
e Extraction of phosphorus—by alkaline ammonium citrate (Petermann’s method) at 65°C
f Extraction of phosphorus—by alkaline ammonium citrate (Petermann’s method) at ambient temperature
g Extraction of phosphorus—by alkaline ammonium citrate (Joulie’s method)
h Extraction of phosphorus—by water
10. Determination of extracted phosphorus
11. Determination of water-soluble potassium
12.a Determination of water-soluble magnesium—atomic absorption spectrophotometric method
b Determination of water-soluble magnesium—EDTA method
13.a Determination of total magnesium—atomic absorption spectrophotometric method
b Determination of total magnesium—EDTA method
14. Determination of chlorides, in the absence of organic matter
15.a Determination of fineness of grinding—dry method
b Determination of fineness of grinding—for soft natural phosphates
16. Methods of analysis and test procedures for ammonium nitrate fertiliser containing more than 28% nitrogen by weight
a Methods for the application of thermal cycles
b Determination of oil retention
c Determination of the combustible ingredients
d Determination of the pH value
e Determination of particle size
f Determination of the chloride content (as chloride ion)
g Determination of copper
1.PREPARATION OF THE SAMPLE FOR ANALYSIS
SCOPE
1. The following procedure is to be used for the preparation of the sample for analysis, taken from the final sample.
PRINCIPLE
2.—2.1 Solid fertilisers: the preparation of a final sample received at the laboratory is a series of operations, usually sieving, grinding and mixing, carriee out in such a way that:—
(a)the smallest amount weighed out laid down by the metthods of analysis is representative of the laboratory sample; and
(b)the fineness of the fertiliser has not been changed by the preparation to the extent that its solubility in the various extraction reagents is appreciably affected.
2.2 Fluid fertilisers: the final sample is mixed by shaking to ensure that any insoluble matter, particularly crystalline material, is thoroughly dispersed before each test portion is taken.
APPARATUS
3.—3.1 Sample divider (optional).
3.2 Sieves with apertures of 0.2 mm and 0.5 mm.
3.3 250 ml flasks, stoppered.
3.4 Porcelain pestle and mortar or grinder.
CHOICE OF TREATMENT TO BE USED
4. Preliminary remark: if the product is suitable, only a representative part of the final sample need be kept.
Final samples which must not be ground
4.1 Calcium nitrate, calcium magnesium nitrate, sodium nitrate, Chile nitrate, calcium cyanamide, nitrogenous calcium cyanamide, ammonium sulphate, ammonium nitrates of over 30% N, urea, basic slag, natural phosphate rendered partially soluble, precipitated dihydrated di-calcium phosphate, calcined phosphate, aluminium calcium phosphate, soft ground rock phosphate.
Final samples which must be divided and part of which must be groud
4.2 These are products in respect of which certain determinations are carried out without previous grinding (fineness of grinding for example) and other determinations after grinding. They include all compound fertilisers containing the following phosphate ingredients: basic slag, aluminium, calcium phosphate, clacined phosphate, soft ground rock phosphate and natural phosphate rendered partially soluble. To that end, divide the final sample into two parts, which are as identical as possible, using a sample divider or by quartering.
Final samples in respect of which all determinations are carried out on a ground product
4.3 These are all other fertilisers on the list which are not to be found under 4.1 and 4.2. The whole final sample shall be ground.
METHOD
5. The part of the final sample referred to under 4.2 and 4.3 is sieved rapidly through a sieve with apertures of 0.5 mm. The residue is ground roughly so as to obtain a product in which there is a minimum of fine particles, and it is then sieved. The grinding must be done in conditions such that the substance is not appreciably heated. The operation is repeated as many times as is necessary until there is not residue, and it must be effected as quickly as possible in order to prevent any gain or loss of constituents (water, ammonia). The whole ground and sieved product is placed in a non-corrodible container provided with an air-tight closure.
Before any weighing is carried out for the analysis, the whole sample must be thoroughly mixed.
SPECIAL CASES
(a) Fertilisers comprising a blend of several categories of crystals
In this case, seperation frequently occurs. It is therefore absolutely essential to crush and pass the sample through a sieve with apertures of 0.2 mm (for example, mixtures of ammonium phosphate and potassium nitrate). The grinding of the whole of the final sample is recommended in the case of these products.
(b) Residue which is difficult to grind and does not contain fertilising substances
Weigh the residue and takle account of its mass when calculating the final result.
(c) Products which decompose on heating
Grinding must be carried out in such a way as to avoid any heating. It is preferable in this case to use a mortar for grinding (for example, compound fertilisers containing calcium cyanamide and urea).
(d) Products which are abnormally moist or made into a paste by grinding
To ensure homogeneity, a sieve is to be chosen which has the smallest apertures compatible with the destruction of lumps by hand or with the pestle. This may be the case of mixtures, certain ingredients of which contain water of crystallisation.
FLUID FERTILISERS
7. Mix thoroughly by shaking, ensuring that any insoluble matter, particularly crystalline material, is thoroughly dispersed, immediately before drawing a portion of the sample of analysis.
2.DETERMINATION OF AMMONIACAL NITROGEN
SCOPE
1. This method is for the determination of ammoniacal nitrogen.
FIELD OF APPLICATION
2. All nitrogeneous fertilisers, including compound fertilisers, in which nitrogen is found exclusively either in the form of ammonium salts, or ammonium salts together with nitrates.
It is not applicable to fertilisers containing urea, cyanamide or other organic nitrogeneous compounds.
PRINCIPLE
3. Displacement of ammonia by means of an excess of sodium hydroxide; distillation; determining the yield of ammonia in a given volume of a standard sulphuric acid and titration of the excess acid by means of a standard solution of sodium or potassium hydroxide.
REAGENTS
4.—4.1 Hydrochloric acid solution, 50% (V/V): dilute an appropriate volume of hydrochloric acid (d= 1.18 g/ml) with an equal volume of water.
4.2 Sulphuric acid, 0.1 N solution. } for variant (a)
4.3 Sodium or potassium hydroxide, 0.1 N solution, carbonate free. } for variant (a)
4.4 Sulphuric acid, 0.2 N solution. } for variant (b) (see Note on page 15)
4.5 Sodium or potassium hydroxide, 0.2 N solution, carbonate free. } for variant (b) (see Note on page 15)
4.6 Sulphuric acid, 0.5 N solution. } for variant (c) (see Note on page 15)
4.7 Sodium or potassium hydroxide, 0.5 N solution, carbonate free. } for variant (c) (see Note on page 15)
4.8 Sodium hydroxide solution, 30g per 100 ml ammonia free.
4.9 Indicator solutions:
4.9.1 Mixed indicator:
4.9.1Solution A: dissolve 1 g methyl red in 37 ml sodium hydroxide solution 0.1 N and make up to 1 litre with water.
Solution B: dissolve 1 g methylene blue in water and make up to 1 litre. Mix 1 volume of solution A and 2 volumes of solution B.
This indicator is violet in acid solution, grey in neutral solution and green in alkaline solution. Use 0.5 ml (10 drops) of this indicator solution.
4.9.2 Methyl red indicator solution:
4.9.2dissolve 0.1 g methyl red in 50 ml ethanol (95%) make up to 100 ml with water and filter if necessary. This indicator may be used (4 to 5 drops) instead of the preceding one.
4.10 Anti-bump granules of pumice stone, washed in hydrochloric acid and ignited.
4.11 Ammonium sulphate.
APPARATUS
5.—5.1 Distillation apparatus consisting of a round-bottomed flask of suitable capacity connected to a condenser by means of a splash head.
Examples of the different types of equipment recommended for this determination are reproduced in Figures 1, 2, 3 and 4 in the Appendix.
5.2 Rotary shaker, 35 to 40 turns per minute.
PREPARATION OF SAMPLE
6. See method 1.
PROCEDURE
7.1.1 Solubility test
Carry out a solubility test on the sample in water at room temperature in the proportion of 2 g per 100 ml.
7.1.2 Preparation of the solution
Weigh to the nearest 0.001 g, according to the indications in the Table, a quantity of 5, 7 or 10 g of the prepared sample and place it in a 500 ml graduated flask. From the result of the solubility test, proceed as follows:
(a)Products completely soluble in water
Add to the flask the quantity of water needed to dissolve the sample; shake, and when completely dissolved, make up the volume and mix thoroughly.
(b)Products not completely soluble in water
Add to the flask 50 ml water and then 20 ml hydrochloric acid solution (4.1). Shake and leave undisturbed until the evolution of carbon dioxide has ceased. Add 400 ml water and shake for half an hour with the rotary shaker (5.2). Make up to volume with water, mix and filter through a dry filter into a dry receiver.
Determination
7.2 According to the variant chosen, place in the collecting flask a measured quantity of standard sulphuric acid as indicated in the table on pages 15 & 16. Add the appropriate quantity of the chosen indicator solution (4.9.1 or 4.9.2) and, if necessary, water in order to obtain a volume of at least 50 ml. The condenser outlet must be below the surface of the standard acid in the collecting flask.
Transfer by pipette, according to the details given in the Table, an aliquot portion of the clear solution into the distillation flask of the apparatus. Add water to obtain a volume of about 350 ml and several grains of pumice in order to control the boiling.
Assemble the distillation apparatus and, taking care to avoid any loss of ammonia, add to the contents of the distillation flask 10 ml of concentrated sodium hydroxide solution (4.8) or 20 ml of the reagent in the cases where 20 ml hydrochloric acid (4.1) have been used in order to dissolve the sample. Warm the flask gently and when boiling commences distil at such rate that about 250 ml are distilled in 30 minutes.
When no more ammonia is likely to be evolved, lower the receving flask so that the tip of the condenser is above the surface of the liquid.
Test the subsequent distillate by means of an appropriate reagent to ensure that all the ammonia is completely distilled. Wash the condenser extension with a little water and titrate the excess acid with the standard solution of sodium or potassium hydroxide prescribed for the variant adopted (see Note).
Note: Standard solutions of different strengths may be used for the titration provided that the volumes used for the titration do not, as far as possible, exceed 40 to 45 ml.
Blank
7.3 Make a blank test under the same conditions (omitting only the sample) and allow for this in the calculation of the final result.
Control test
7.4 Before carrying out analyses, check that the apparatus is working properly and that the correct application of the method is used, using an aliquot part of a freshly prepared solution of ammonium sulphate (4.11) containing the maximum quantity of nitrogen prescribed for the chosen variant.
EXPRESSION OF THE RESULT
8. Express the result of the analysis as the percentage of ammoniacal nitrogen in the fertiliser as received for analysis.
TABLE FOR METHOD 2
Determination of the ammoniacal nitrogen and of the ammoniacal and nitrate nitrogen in fertilisers.
Table of the weighing, dilution and calculation to be carried out for each of the variants (a), (b) and (c) of the method.
Variant (a)—
Approximate maximum quantity of nitrogen to be distilled = 50 mg
Sulphuric acid 0.1 N to be placed in the receiving flask = 50 ml
Titration with sodium or potassium hydroxide, 0.1 N solution
Declaration N% Amount to be weighed (g) Dilution (ml) Solution of sample to be distilled (ml) Expression of the result (1) N% = (50 − A) F (1)For the purposes of the formula for expression of the result:
50 or 35=
millilitres of standard solution of sulphuric acid to be placed in the receiving flask.
A=
millilitres of sodium or potassium hydroxide used for the titration.
F=
factor taking into account the weight of sample, the dilution, the volume of the aliquot part distilled and the volumetric equivalent.
0 — 5 10 500 50 (50 − A) × 0.14 5 — 10 10 500 25 (50 − A) × 0.28 10 — 15 7 500 25 (50 − A) × 0.40 15 — 20 5 500 25 (50 − A) × 0.56 20 — 40 7 500 10 (50 − A) × 1.00
Variant (b)—
Approximate maximum quantity of nitrogen to be distilled = 100 mg
Sulphuric acid 0.2 N to be placed in the receiving flask = 50 ml
Titration with sodium or potassium hydroxide, 0.2 N solution
Declaration N% Amount to be weighed (g) Dilution (ml) Solution of sample to be distilled (ml) Expression of the result (1) N% = (50 − A) F (1)For the purposes of the formula for expression of the result:
50 or 35=
millilitres of standard solution of sulphuric acid to be placed in the receiving flask.
A=
millilitres of sodium or potassium hydroxide used for the titration.
F=
factor taking into account the weight of sample, the dilution, the volume of the aliquot part distilled and the volumetric equivalent.
0 — 5 10 500 100 (50 − A) × 0.14 5 — 10 10 500 50 (50 − A) × 0.28 10 — 15 7 500 50 (50 − A) × 0.40 15 — 20 5 500 50 (50 − A) × 0.56 20 — 40 7 500 50 (50 − A) × 1.00
Variant (c)—
Approximate mavimum quantity of nitrogen to be distilled = 200 mg
Sulphuric acid 0.5 N to be placed in the receiving flask = 35 ml
Titration with sodium or potassium hydroxide, 0.5 N solution
Declaration N% Amount to be weighed (g) Dilution (ml) Solution of sample to be distilled (ml) Expression of the result (1) N% = (35 − A) F (1)For the purposes of the formula for expression of the result: 50 or 35 = millilitres of standard solution of sulphuric acid to be placed in the receiving flask.
A=
millilitres of sodium or potassium hydroxide used for the titration.
F=
factor taking into account the weight of sample, the dilution, the volume of the aliquot part distilled and the volumetric equivalent.
0 — 5 10 500 200 (35 − A) × 0.175 5 — 10 10 500 100 (35 − A) × 0.350 10 — 15 7 500 100 (35 − A) × 0.500 15 — 20 5 500 100 (35 − A) × 0.700 20 — 40 5 500 50 (35 − A) × 1.400
3a.DETERMINATION OF NITRIC AND AMMONIACAL NITROGEN— ULSCH METHOD
SCOPE
1. This method is for the determination of nitric and ammoniacal nitrogen with reduction according to Ulsch.
FIELD OF APPLICATION
2. All nitrogeneous fertilisers, including compound fertilisers, in which nitrogen is found exclusively in nitrate form, or in ammoniacal and nitrate form.
PRINCIPLE
3. Reduction of nitrates and nitrites to ammonia by means of metallic iron in an acid medium and displacement of the ammonia thus formed by the addition of an excess of sodium hydroxide: distillation of the ammonia and determination of the yield of ammonia in a known volume of standard sulphuric acid solution. Titration of the excess sulphuric acid by means of a standard solution of sodium or potassium hydroxide.
REAGENTS
4.—4.1 Hydrochloric acid solution, 50% (V/V): dilute an appropriate volume of hydrochloric acid (d = 1.18 g/ml) with an equal volume of water.
4.2 Sulphuric acid, 0.1 N solution.
4.3 Sodium or potassium hydroxide, 0.1 N solution, carbonate free.
4.4 Sulphuric acid solution, approximately 30%H2SO4 (W/V), ammonia free.
4.5 Powdered iron reduced in hydrogen. (The prescribed quantity of iron must be able to reduce at least 0.05 nitrate nitrogen.)
4.6 Sodium hydroxide solution, 30 g per 100 ml, ammonia free.
4.7 Indicator solutions:
4.7.1 Mixed indicator:
4.7.1Solution A: dissolve 1 g methyl red in 37 ml 0.1 N sodium hydroxide solution and make up to 1 litre with water.
Solution B: dissolve 1 g methylene blue in water and make up to 1 litre.
Mix 1 volume of solution A with 2 volumes of solution B.
This indicator is violet in acid solution, grey in neutral solution and green in alkaline solution: use 0.5 ml (10 drops).
4.7.2 Methyl red indicator solution:
4.7.2dissolve 0.1 g methyl red in 50 ml 95% ethanol, make up to 100 ml with water and filter if necessary.
This indicator may be used (4 — 5 drops) instead of the preceding one.
4.8 Anti-bump granules of pumice stone, washed in hydrochloric acid and ignited.
4.9 Sodium nitrate.
APPARATUS
5. See method 2.
PREPARATION OF SAMPLE
6. See method 1.
PROCEDURE
Preparation of the solution
7.—7.1 See Method 2.
Determination
7.2 Place in the receiving flask an exactly measured quantity of standard sulphuric acid (4.2) as indicated in the Table of Method 2 (variant (a)) and add the appropriate quantity of indicator solution (4.7.1 or 4.7.2).
The end of the extension tube of the condenser must be below the surface of the standard acid in the receiving flask.
Using a pipette, transfer an aliquot part of the clear solution as indicated in the Table of Method 2 (variant (a)) and place it in the distilling flask of the apparatus. Add 350 ml water, 20 ml 30% sulphuric acid solution (4.4), stir, and add 5 g of reduced iron (4.5). Wash the neck of the flask with several ml water, and place in the neck of the flask a small, long stemmed funnel. Heat in a boiling water bath for an hour and then wash the stem of the funnel with a few ml of water.
Taking care to avoid any loss of ammonia, add to the contents of the distilling flask 50 ml concentrated sodium hydroxide solution (4.6), or in the cases where 20 ml of hydrochloric acid (4.1) has been used to dissolve the sample, add 60 ml of concentrated sodium hydroxide solution (4.6). Assemble the distillation apparatus. Distil the ammonia according to the procedure given in Method 2. Titrate the excess acid with the standard solution of sodium or potassium hydroxide (4.3).
Blank test
7.3 Carry out a blank test (omitting only the sample) under the same conditions and allow for this in the calculation of the final result.
Control test
7.4 Before analysis check that the apparatus is working properly and that the correct application of the method is used by using an aliquot part of a freshly prepared solution of sodium nitrate (4.9) containing 0.045 g to 0.050 g of nitrogen.
EXPRESSION OF THE RESULTS
8. Express the results of analysis as a percentage of nitric nitrogen or combined ammoniacal and nitric nitrogen contained in the fertiliser as received for analysis.
3b.DETERMINATION OF NITRIC AND AMMONIACAL NITROGEN—ARND METHOD
SCOPE
1. This method is for the determination of nitric and ammoniacal nitrogen with reduction according to Arnd (modified for each of the variants (a), (b) and (c)).
FIELD OF APPLICATION
2. See method 3a.
PRINCIPLE
3. Reduction of nitrates and nitrites to ammonia in a neutral aqueous solution by means of metallic alloy composed of 60% Cu and 40% Mg (Arnd’s alloy) in the presence of magnesium chloride.
Distillation of the ammonia and determination of the yield in a known volume of standard sulphuric acid solution. Titration of the excess acid by means of a standard solution of sodium or potassium hydroxide.
REAGENTS
4.—4.1 Hydrochloric acid solution, 50% (V/V): dilute an appropriate volume of hydrochloric acid (d = 1.18 g/ml) with an equal volume of water.
4.2 Sulphuric acid, 0.1 N solution. } for variant (a) (page 15)
4.3 Sodium or potassium hydroxide, 0.1 N solution, carbonate free. } for variant (a) (page 15)
4.4 Sulphuric acid, 0.2 N solution. } for variant (b) page (16) (see Note on page 15)
4.5 Sodium or potassium hydroxide, 0.2 N solution, carbonate free. } for variant (b) page (16) (see Note on page 15)
4.6 Sulphuric acid, 0.5 N solution. } for variant (c) (page 16) (see Note on page 15)
4.7 Sodium or potassium hydroxide, 0.5 N solution, carbonate free. } for variant (c) (page 16) (see Note on page 15)
4.8 Sodium hydroxide solution, approximately 2 N.
4.9 Arnd’s alloy—powdered so as to pass through a sieve with apertures less than 1.00 mm square.
20% Magnersium chloride solution:
4.10 dissolve 200 g magnesium chloride (MgCL2.6H20) in approximately 600-700 ml water in a litre flat-bottomed flask. To prevent frothing, add 15 g magnesium sulphate (MgSO4.7H2O). After dissolution add 2 g magnesium oxide and a few anti-bump granules of pumice stone and concentrate the suspension to 200 ml by boiling, thus expelling any trace of ammonia from the reagents. Cool, make up the volume to 1 litre and filter.
Indicator solutions
4.11.1 Mixed indicator:
4.11.1Solution A: dissolve 1 g methyl red in 37 ml 0.1 N sodium hydroxide solution and make up to 1 litre with water.
Solution B: dissolve 1 g methylene blue in water and make up to 1 litre. Mix 1 volume of A with 2 volumes of B.
This indicator is violet in acid solution, grey in neutral solution and green in alkaline solution.
Use 0.5 ml (10 drops).
4.11.2 Methyl red indicator solution:
4.11.2dissolve 0.1 g methyl red in 50 ml 95% ethanol, make up to 100 ml with water and filter if necessary. This indicator may be used (4 to 5 drops) instead of the preceding one.
4.11.3 Congo red indicator solution:
4.11.3dissolve 3 g Congo red in 1 litre warm water and filter if necessary after cooling. This indicator may be used, instead of the two described above, in the neutralisation of acid extracts before distillation, using 0.5 ml per 100 ml of liquid to be neutralised.
4.12 Anti-bump granules of pumice stone wahed in hydrochloric acid and ignited.
4.13 Sodium nitrate.
APPARATUS
5. See method 2.
PREPARATION OF SAMPLE
6. See method 1.
PROCEDURE
Preparation of the solution for analysis
7.—7.1 See method 2.
Determination
7.2 According to the chosen variant, place in the receiving flask a measured quantity of standard sulphuric acid as indicated in the Table of Method 2. Add the appropriate quantity of chosen indicator solution (4.11.1 or 4.11.2) and if necessary water to give a volume of at least 50 ml. The end of the extension tube of the condenser must be below the surface of the solution.
Using a pipette, take, according to the Table, an aliquot part of the clear solution and place in the distillation flask.
Add sufficient water to obtain a total volume of about 350 ml (see Note), 10 g Arnd’s alloy (4.8), 50 ml magnesium chloride solution (4.10) and a few fragments of pumice stone (4.12). Rapidly connect the flask to the distillation apparatus. Heat gently for about 30 minutes. Then increase the heating to distil the ammonia. Continue the ditillation for about an hour.
After this time, the residue in the flask ought to have a syrupy consistency. When the distillation has finished, titrate the quantity of excess acid in the receiving flask according to the procedure in Method 2.
Note: When the sample solution is acid (addition of 20 ml hydrochloric acid (4.1) to dissolve the sample) the aliquot part taken for analysis is neutralised in the following way:
to the distillation flask containing the aliquot part add about 250 ml water, the necessary quantity of one of the indicators (4.11.1, 4.11.2, 4.11.3) and shake carefully.
Neutralise with 2 N sodium hydroxide solution (4.8) and acidify again with a drop of hydrochloric acid (4.1). The proceed as indicated in 7.2.
Blank test
7.3 Carry out a blank test under the same conditions (omitting only the sample) and allow for this in the calculation of the final result.
Control test
7.4 Before analysis, check that apparatus is working properly and that the correct technique is applied using a freshly prepared solution of sodium nitrate (4.13) containing 0.050 g to 0.150 g nitrogen depending on the variant chosen.
EXPRESSION OF THE RESULTS
8. Express the results of analysis as a percentage of nitric nitrogen or combined ammoniacal and nitric nitrogen contained in the fertiliser as received for analysis.
3c.DETERMINATION OF NITRIC AND AMMONIACAL NITROGEN—DEVARDA METHOD
SCOPE
1. This method is for the determination of nitric and ammoniacal nitrogen with reduction according to Devarda (modified for each of the variants (a), (b) and (c)).
FIELD OF APPLICATION
2. See Method 3a.
PRINCIPLE
3. Reduction of nitrates and nitrites to ammonia in a strongly alkaline solution by means of a metallic alloy composed of 45% Al, 5% Zn and 50% Cu (Devarda alloy). Distillation of the ammonia and determination of the yield in a known volume of standard sulphuric acid; titration of the excess sulphuric acid by means of a standard solution of sodium or potassium hydroxide.
REAGENTS
4.—4.1 Hydrochloric acid solution, 50% (V/V): dilute an appropriate volume of hydrochloric acid (d = 1.18 g/ml) with an equal volume of water.
4.2 Sulphuric acid, 0.1 N solution. } for variant (a) (page 15)
4.3 Sodium or potassium hydroxide, 0.1 N solution, carbonate free. } for variant (a) (page 15)
4.4 Sulphuric acid, 0.2 N solution. } for variant (b) (page 16) (see Note on page 15)
4.5 Sodium or potassium hydroxide, 0.2 N solution, carbonate free. } for variant (b) (page 16) (see Note on page 15)
4.6 Sulphuric acid, 0.5 N solution. } for variant (c) (page 16) (see Note on page 15)
4.7 Sodium or potassium hydroxide, 0.5 N solution, carbonate free. } for variant (c) (page 16) (see Note on page 15)
4.8 Devarda’s alloy—powdered so that 90 to 100% will pass through a sieve with apertures less than 0.25 mm square, 50 to 75% will pass through a sieve with apertures of less than 0.075 mm square. (Pre-packed bottles containing a maximum of 100 g are recommended.)
4.9 Sodium hydroxide solution, 30 g per 100 ml ammonia free.
4.10 Indicator solutions:
4.10.1 Mixed indicator:
4.10.1Solution A: dissolve 1 g methyl red in 37 ml 0.1 N sodium hydroxide solution and make up to 1 litre with water.
Solution B: dissolve 1 g methylene blue in water and make up to 1 litre. Mix 1 volume of A with 2 volumes of B.
This indicator is violet in acid solution, grey in neutral solution and green in alkaline solution.
Use 0.5 ml (10 drops).
4.10.2 Methyl red indicator:
4.10.2dissolve 0.1 g methyl red in 50 ml 95% ethanol. Make up to 100 ml with water and filter if necessary.
This indicator (4 to 5 drops) may be used instead of the preceding one.
4.11 Ethanol, 95%.
4.12 Sodium nitrate.
APPARATUS
5.—5.1 Distillation apparatus consisting of a round bottomed flask of suitable capacity, connected to a condenser by means of a splash head, equipped, in addition, with a bubble trap on the receiving flask to prevent any loss of ammonia.
An example of the type of apparatus recommended for this determination is reproduced in Figure 5 in the Appendix.
PREPARATION OF THE SAMPLE
6. See Method 1.
PROCEDURE
Preparation of the solution for analysis
7.—7.1 See Method 2.
Determination
7.2 According to the variant chosen, place in the receiving flask an exactly measured quantity of standard sulphuric acid as indicated in the Table. Add the appropriate quantity of the chosen indicator solution (4.10.1 or 4.10.2) and finally, sufficient water to give a volume of 50 ml. The end of the extension tube of the condenser must be below the surface of the solution. Fill the bubble trap with distilled water.
Using a pipette, take an aliquot part of the clear solution as indicated in the Table and place in the distillation flask. Add sufficient water to the distillation flask to obtain a volume of 250-300 ml, 5 ml ethanol (4.11) and 4 g Devarda’s alloy (4.8).
(Note: In the presence of calcium salts such as calcium nitrate and calcium ammonium nitrate, it is necessary to add, before distillation for each gram of sample present in the aliquot, 0.700 g disodium hydrogen phosphate (Na2HPO4.2H2O) to prevent the formation of calcium hydroxide.)
Taking the necessary precautions to avoid loss of ammonia, add to the flask about 30 ml 30% sodium hydroxide solution (4.9) and finally, in the case of acid soluble samples, an additional quantity sufficient to neutralise the quantity of hydrochloric acid (4.1) present in the aliquot part taken for the analysis. Connect the distillation flask to the apparatus, ensuring the tightness of connections. Carefully shake the flask to mix the contents.
Warm gently, so that the release of hydrogen decreases appreciably over about half an hour and then liquid will boil. Continue the distillation, increasing the heat so that at least 200 ml liquid distils in about 30 minutes. (Do not prolong the distillation beyond 45 minutes.)
When the distillation is complete, disconnect the receiving flask from the apparatus, carefully wash the extension tube and bubble trap, collecting the rinsings in the titration flask. Titrate the excess acid according to the procedure in Method 2.
Blank test
7.3 Carry out a blank test under the same conditions omitting only the sample and allow for this in the calculation of the final results.
Control test
7.4 Before carrying out the analysis, check that the apparatus is working properly and that the correct application of the method is used, using an aliquot of a freshly prepared solutionj of sodium nitrate (4.12) containing, according to the variant chosen, 0.050 g to 0.150 g nitrate nitrogen.
EXPRESSION OF RESULTS
8. Express the results of analysis as a percentage of nitric nitrogen or combined ammoniacal and nitric nitrogen contained in the fertiliser as received for analysis.
4a.DETERMINATION OF THE TOTAL NITROGEN IN CALCIUM CYANAMIDE—IN THE ABSENCE OF NITRATE
SCOPE
1. This method is for the determination of total nitrogen in nitrate free calcium cyanamide.
FIELD OF APPLICATION
2. Exclusively to calcium cyanamide (nitrate free).
PRINCIPLE
3. After Kjeldahl digestion, the ammoniacal nitrogen formed is displaced by sodium hydroxide and collected in a standard solution of sulphuric acid. The excess sulphuric acid is titrated with a standard solution of sodium or potassium hydroxide.
REAGENTS
4.—4.1 Sulphuric acid solution 50% (V/V): dilute an appropriate volume of sulphuric acid (d = 1.84 g/ml) with an equal volume of water.
4.2 Potassium sulphate.
4.3 Copper oxide (CuO)—0.3 to 0.4 g for each determination or an equivalent quantity of copper sulphate pentahydrate, from 0.95 to 1.25 g for each determination.
4.4 Sodium hydroxide solution, 30 g per 100 ml, ammonia free.
4.5 Sulphuric acid, 01. N solution. } for variant (a) (page 15)
4.6 Sodium or potassium hydroxide, o.1 N solution, carbonate free. } for variant (a) (page 15)
4.7 Sulphuric acid, 0.2 N solution. } for variant (b) (page 16) (see Note on page 15)
4.8 Sodium or potassium hydroxide, 0.2 N solution, carbonate free. } for variant (b) (page 16) (see Note on page 15)
4.9 Sulphuric acid, 0.5 N solution. } for variant (c) (page 16) (see Note on page 15)
4.10 Sodium or potassium hydroxide, 0.5 N solution, carbonate free. } for variant (c) (page 16) (see Note on page 15)
4.11 Indicator solutions:
4.11.1 Mixed indicator:
4.11.1Solution A: dissolve 1 g methyl red in 37 ml 0.1 N sodium hydroxide solution and make up to 1 litre with water.
Solution B: dissolve 1 g methylene blue in water and make up to 1 litre.
Mix 1 volume of A with 2 volumes of B.
This indicator is violet in acid solution, grey in neutral solution and green in alkaline solution.
Use 0.5 ml (10 drops).
4.11.2 Methyl red indicator:
4.11.2dissolve 0.1 g methyl red in 50 ml 95% ethanol and make up to 100 ml with water. Filter if necessary. This indicator (4 to 5 drops) may be used instead of the preceding one.
4.12 Anti-bump granules of pumice stone, washed in hydrochloric acid and ignited.
4.13 Potassium thiocyanate.
APPARATUS
5.—5.1 Distillation apparatus. See Method 2.
PREPARATION OF SAMPLE
6. See Method 1.
PROCEDURE
Preparation of the solution
7.—7.1 Weigh to the nearest 0.001 g, 1 g of the prepared sample and place it in the Kjeldahl flask. Add 50 ml 50% sulphuric acid (4.1), 10-15 g potassium sulphate (4.2) and one of the prescribed catalysts (4.3). Heat slowly to drive off the water, boil gently for two hours, allow to cool, and dilute with 100-150 ml water. Cool again, transfer quantitatively the suspension to a 250 ml graduated flask, make up to volume with water, shake and filter through a dry filter into a dry flask.
Determination
7.2 According to the variant chosen (see Method 2) transfer with a pipette 50, 100 or 200 ml of the solution to the distillation apparatus and add sufficient sodium hydroxide solution (4.4) to ensure a considerable excess. Distil the ammonia and titrate the excess acid as described in Method 2.
Blank test
7.3 Make a blank test (omitting only the sample ) under the same conditions and allow for this in the calculation of the final result.
Control test
7.4 Before carrying out the analysis, check that the apparatus is working properly and that the correct application of the method is used, using an aliquot part of a standard solution of potassium thiocyanate (4.13), approximating to the concentration of nitrogen in the sample.
EXPRESSION OF THE RESULT
8. Express the result as the percentage of nitrogen (N) contained in the fertiliser as received for analysis.
Variant (a): N% = (50 − A) × 0.7
Variant (b): N% = (50 − A) × 0.7
Variant (c): N% = (35 − A) × 0.875
Where A = millilitres of sodium or potassium hydroxide used for the titration.
4b.DETERMINATION OF TOTAL NITROGEN IN CALCIUM CYANAMIDE—IN THE PRESENCE OF NITRATE
SCOPE
1. This method is for the determination of total nitrogen in calcium cyanamide.
FIELD OF APPLICATION
2. The method is applicable to calcium cyanamide containing nitrates.
PRINCIPLE
3. The direct application of Kjeldahl’s method cannot be applied to calcium cyanamides containing nitrates. For this reason the nitric nitrogen is reduced to ammonia with mewtallic iron and stannous chloride before Kjeldahl digestion. The ammoniacal nitrogen is that determined as in Method 4a.
REAGENTS
4.—4.1 Sulphuric acid (d = 1.84 g/ml).
4.2 Powdered iron reduced in hydrogen.
4.3 Potassium sulphate, finely pulverised.
4.4 Sulphuric acid, 0.1 N solution. } for variant (a) (page 15)
4.5 Sodium or potassium hydroxide, 0.1 N solution, carbonate free. } for variant (a) (page 15)
4.6 Suplhuric acid, 0.2 N solution. } for variant (b) (page 16) (see Note on page 15)
4.7 Sodium or potassium hydroxide, 0.2 N solution, carbonate free. } for variant (b) (page 16) (see Note on page 15)
4.8 Sulphuric acid, 0.5 N solution. } for variant (c) (page 16) (see Note on page 15)
4.9 Sodium or potassium hydroxide, 0.5 N solution, carbonate free. } for variant (c) (page 16) (see Note on page 15)
4.10 Indicator solutions:
4.10.1 Mixed indicator:
4.10.1Solution A: dissolve 1 g of methyl red in 37 ml of the 0.1 N sodium hydroxide solution and make up to 1 litre with water.
Solution B: dissolve 1 g of methylene blue in water and make up to 1 litre. Mix 1 volume of A and 2 volumes of B.
This indicator is violet in acid solution, grey in neutral solution and green in alkaline solution.
Take 0.5 ml (10 drops) of this indicator solution.
4.10.2 Methyl red indicator:
4.10.2dissolve 0.1 g of methyl red in 50 ml of 95% ethanol, make up to 100 ml with water and filter if necessary. This indicator (4 to 5 drops) may be used instead of the preceding one.
Solution of stannous chloride:
4.11 Dissolve 120 g of stannous chloride (SnCl2.2H2O) in 400 ml concentrated hydrochloric acid (d = 1.18 g/ml) and make up to 1 litre with water. The solution must be completely clear and prepared immediately before use. It is essential to check the reducing power of the stannous chloride. Dissolve 0.5 g of stannous chloride in 2 ml concentrated hydrochloric acid (d = 1.18 g/ml) and make up to 50 ml with water. Then add 5 g of Rochelle salt (potassium sodium tartrate) and a sufficient quantity of sodium bicarbonate for the solution to show an alkaline reaction to a litmus paper test.
Titrate with 0.1 N iodine solution in the presence of a starch solution as an indicator.
1 ml of iodine solution 0.1 N corresponds to 0.01128 g SnCl2.2H2O.
At least 80% of the total tin present in the solution thus prepared must be in a bivalent form. For the titration at least 35 ml of 0.1 N iodine solution should be used.
4.12 Sodium hydroxide solution, 30 g per 100 ml, ammonia free.
Standard nitrate-ammoniacal solution:
4.13 Weigh out 2.500 g of potassium nitrate and 10.160 g of ammonium sulphate and place them in a 250 ml graduated flask. Dissolve in water and make up to 250 ml. 1 ml of this solution contains 0.010 g of nitrogen.
4.14 Anti-bump granules of pumice stone, washed in hydrochloric acid and ignited.
APPARATUS
5. Distillation apparatus. See Method 2.
PREPARATION OF THE SAMPLE
6. See Method 1.
PROCEDURE
Preparation of the solution
7.—7.1 Weigh to the nearest 0.001 g, 1 g of the prepared sample and place in the Kjeldahl flask. Add 0.5 g of powdered iron (4.2) and 50 ml of the stannous chloride solution (4.11), stir and leave standing for half an hour. During this time it is left standing, stir again after 10 and 20 minutes. Then add 10 g of potassium sulphate (4.3) and 30 ml of sulphuric acid (4.1). Boil and carry on the process for an hour after the appearance of white fumes. Leave to cool and dilute with 100 — 150 ml of water. Transfer the suspension quantitatively into a 250 ml graduated flask, cool and make up to volume with water, mix and filter through a dry filter into a dry container.
Determination
7.2 According to the variant chosen (see Method 2) transfer with a pipette 50, 100 or 200 ml of the solution to the distillation apparatus and add sufficient sodium hydroxide solution (4.12) to ensure a considerable excess. Distil the ammonia and titrate the excess acid as described in Method 2.
Blank test
7.3 Make a blank test (omitting only the sample) under the same conditions and allow for this in the calculation of the final result.
Control test
7.4 Before carrying out the analysis, check that the apparatus is working properly and that the correct application of the method is used with a standard solution containing quantities of ammoniacal and nitrate nitrogen comparable to the quantities of cyanamide and nitrate nitrogen contained in nitrated calcium cyanamide.
For this purpose place 20 ml of the standard solution (4.13) in the Kjeldahl flask.
Carry out the analysis according to the method described in paragraph 7.
EXPRESSION OF THE RESULTS
8. The result of the analysis must be expressed as the percentage of total nitrogen (N) contained in the fertiliser as received for analysis.
Variant (a): N% = (50 − A) × 0.7
Variant (b): N% = (50 − A) × 0.7
Variant (c): N% = (35 − A) × 0.875
Where A = millilitres of sodium or potassium hydroxide used for the titration.
5.DETERMINATION OF TOTAL NITROGEN IN UREA
SCOPE
1. This method is for the determination of total nitrogen in urea.
FIELD OF APPLICATION
2. This method is applied exclusively to urea fertilisers which are nitrate free.
PRINCIPLE
3. Urea is transformed quantitatively into ammonia by boiling in the presence of sulphuric acid. The ammonia thus obtained is distilled from an alkaline medium and collected in an excess of standard sulphuric acid. The excess acid is titrated by means of a standard alkaline solution.
REAGENTS
4.—4.1 Sulphuric acid, concentrated, (d = 1.84 g/ml).
4.2 Sodium hydroxide solution, 30 g per 100 ml, ammonia free.
4.3 Sulphuric acid, 0.1 N solution. } for variant (a) (page 15)
4.4 Sodium or potassium hydroxide, 0.1 N solution, carbonate free. } for variant (a) (page 15)
4.5 Sulphuric acid, 0.2 N solution. } for variant (b) (page 16) (see Note on page 15)
4.6 Sodium or potassium hydroxide, 0.2 N solution, carbonate free. } for variant (b) (page 16) (see Note on page 15)
4.7 Sulphuric acid, 0.5 N solution. } for variant (c) (page 16) (see Note on page 15)
4.8 Sodium or potassium hydroxide, 0.5 N solution, carbonate free. } for variant (c) (page 16) (see Note on page 15)
4.9 Indicator solutions:
4.9.1 Mixed indicator:
4.9.1Solution A: dissolve 1 g methyl red in 37 ml 0.1 N sodium hydroxide solution and make up to 1 litre with water.
Solution B: dissolve 1 g methylene blue in water and make up to 1 litre.
Mix 1 volume of solution A with 2 volumes of solution B.
This indicator is violet in acid solution, grey in neutral solution and green in alkaline solution.
Use 0.5 ml (10 drops).
4.9.2 Methyl red indicator solution:
4.9.2dissolve 0.1 g methyl red in 50 ml 95% ethanol and make up to 100 ml with water. Filter if necessary. This indicator (4 — 5 drops) may be used instead of the preceding one.
4.10 Anti-bump granules of pumice stone, washed in hydrochloric acid and ignited.
4.11 Urea.
APPARATUS
5.—5.1 Distillation apparatus. See Method 2.
PREPARATION OF THE SAMPLE
6. See Method 1.
PROCEDURE
Preparation of the solution
7.—7.1 Weigh to the nearest 0.001 g, 2.5 g of the prepared sample, place in a 300 ml Kjeldahl flask and moisten with 20 ml water. Stir in 20 ml concentrated sulphuric acid (4.1) and add a few glass beads to prevent bumping. To prevent splashing, place a long-stemmed glass funnel in the neck of the flask. Heat slowly at first, then increase the heat until white fumes are observed (30 — 40 minutes).
Cool and dilute with 100 — 150 ml water. Quantitatively transfer to a 500 ml graduated flask, discarding any sediment. Allow to cool to room temperature. Make up to volume with water, mix and, if necessary, filter through a dry filter into a dry receptacle.
Determination
7.2 According to the variant chosen (see Method 2) transfer with a pipette 25, 50 or 100 ml of the solution to the distillation apparatus and add sufficient sodium hydroxide solution (4.2) to ensure a considerable excess. Distil the ammonia and titrate the excess acid as described in Method 2.
Blank test
7.3 Carry out a blank test (omitting only the sample) under the same conditions and allow for this in the calculation of the final result.
Control test
7.4 Before carrying out the analysis, check that the apparatus is working properly and that the correct application of the method is used, using an aliquot part of a freshly prepared solution of urea (4.11).
EXPRESSION OF THE RESULT
8. Express the result as the percentage of nitrogen (N) contained in the fertiliser as received for analysis.
Variant (a): N% = (50 − A) × 1.12
Variant (b): N% = (50 − A) × 1.12
Variant (c): N% = (35 − A) × 1.40
6.DETERMINATION OF CYANAMIDE NITROGEN
SCOPE
1. This method is for the determination of cyanamide nitrogen.
FIELD OF APPLICATION
2. Calcium cyanamide and calcium cyanamide/nitrate mixtures.
PRINCIPLE
3. Cyanamide nitrogen is precipitated as a silver complex and estimated in the precipitate by Kjeldahl’s method.
REAGENTS
4.—4.1 Glacial acetic acid.
4.2 Ammonia solution: dilute on volume of ammonia (d = 0.88 g/ml) with 3 volumes of water.
4.3 Ammoniacal silver solution, according to Tollens, freshly prepared: mix 500 ml silver nitrate solution (10 g per 100 ml) with 500 ml ammonia solution (4.2).
Do not expose unnecessarily to light, heat or air.
Safety precaution: when handling ammoniacal silver nitrate solution, safety goggles must be worn.
4.4 Concentrated sulphuric acid (d = 1.84 g/ml).
4.5 Potassium sulphate.
4.6 Copper oxide (CuO), 0.3 — 0.4 g for each determination or an equivalent quantity of copper sulphate pentahydrate (0.95 — 1.25 g) for each determination.
4.7 Sodium hydroxide solution, 30 g per 100 ml, ammonia free.
4.8 Sulphuric acid, 0.1 N solution.
4.9 Sodium or potassium hydroxide, 0.1 N solution.
4.10 Indicator solutions:
4.10.1 Mixed indicator:
4.10.1Solution A: dissolve 1 g methyl red in 37 ml 0.1 N sodium hydroxide solution and make up to 1 litre with water.
Solution B: dissolve 1 g methylene blue in water and make up to 1 litre.
Mix 1 volume of solution A with 2 volumes of solution B.
This indicator is violet in acid solution, grey in neutral solution and green in alkaline solution.
Use 0.5 ml (10 drops).
4.10.2 Methyl red indicator solution:
4.10.2dissolve 0.1 g methyl red in 50 ml 95% ethanol and make up to 100 ml with water. Filter if necessary. This indicator (4 to 5 drops) may be used instead of the preceding one.
4.11 Anti-bump granules of pumice stone, washed in hydrochloric acid and ignited.
4.12 Potassium thiocyanate.
APPARATUS
5.—5.1 Distillation apparatus. See Method 2.
5.2 500 ml graduated flask (for example Stohmann).
5.3 Rotary shaker, 35 — 40 turns per minute.
PREPARATION OF THE SAMPLE
6. See Method 1.
PROCEDURE
Safety precaution
7.—7.1 When handling any ammoniacal silver nitrate solution safety goggles must be worn.
Preparation of the solution for analysis
7.2 Weigh to the nearest 0.001 g, 2.5 g of the prepared sample and place in a small glass mortar. Grind the sample three times with water, pouring of the water after each grinding into the 500 ml graduated flask (5.2). Transfer quantitatively the sample into the flask, washing the mortar, pestle and funnel with water. Make up with water to approximately 400 ml. Add 15 ml acetic acid (4.1). Shake on the rotary shaker (5.5) for two hours.
Make up to 500 ml with water, mix and filter.
Proceed immediately to 7.3
Determination
7.3 Transfer 50.0 ml of the filtrate into a 250 ml beaker. Add ammonia solution (4.2) until slightly alkaline and add 30 ml warm ammoniacal silver nitrate (4.3) in order to precipitate the yellow silver compex of the cyanamide. Leave overnight, filter and wash the precipitate with cold water until completely free of ammonia.
Place the filter and precipitate, still moist, in a Kjeldahl flask, add 10 — 15 g potassium sulphate (4.5), the catalyst (4.6) in the prescribed proportion, then 50 ml water and 25 ml concentrated sulphuric acid (4.4). Warm the flask slowly, whilst shaking it gently until the contents come to the boil. Increase the heat, boil until the contents of the flask become either colourless or pale green. Continue boiling for one hour, then leave to cool.
Transfer the liquid quantitatively from the Kjeldahl flask to the distilling flask, add a few anti-bump granules of pumice stone (4.11) and make up with water to a total volume of approximately 350 ml. Mix and cool. Add sufficient sodium hydroxide solution (4.7) to ensure a considerable excess.
Distil the ammonia and titrate the excess acid as described in Method 2 (variant (a)).
Blank test
7.4 Make a blank test (omitting only the sample) under the same conditions and allow for this in the calculation of the final result.
Control test
7.5 Before carrying out the analysis, check that the apparatus is working properly and that the correct application of the method is used, using an aliquot part of a standard solution of potassium thiocyanate (4.12), corresponding to 0.05 g of nitrogen.
EXPRESSION OF RESULTS
8. Express the result as the percentage of cyanamide nitrogen contained in the fertiliser as received for analysis.
N% = (50 − A) × 0.56
Where A = millilitres of sodium or potassium hydroxide used for the titration.
7.DETERMINATION OF BIURET IN UREA
SCOPE
1. This method is for the determination of biuret in urea.
FIELD OF APPLICATION
2. The method is applied exclusively to urea.
PRINCIPLE
3. In an alkaline medium, in the presence of potassium sodium tartrate, biuret and bivalent copper from a violet cupric compound, the absorbance of which is measured at 546 mm.
REAGENTS
4.—4.1 Methanol.
4.2 Sulphuric acid solution, approximately 0.1 N.
4.3 Sodium hydroxide solution, approximately 0.1 N.
4.4 Alkaline solution of potassium sodium tartrate:
In a 1 litre graduated flask dissolve 40 g of sodium hydroxide in 500 ml of water and leave to cool. Add 50 g of potassium sodium tartrate (KNaC4H4O6.4H2O). Make up to the mark and mix. Leave standing 24 hours before use.
Copper suplhate solution:
4.5 In a litre graduated flask dissolve 15 g of copper sulphate (CuSO4.5H2O) in 500 ml of water.
Make up to the mark and mix.
Biuret standard solution:
4.6 In a 250 ml graduated flask, dissolve 0.250 g of pure biuret(13) in water. Make up to the mark and mix. 1 ml of this solution contains 0.001 g of biuret. This solution should be freshly prepared.
Methyl red indicator solution:
4.7 Dissolve 0.1 g methyl red in 50 ml 95% ethanol and make up to 100 ml with water. Filter if necessary.
APPARATUS
5.—5.1 Spectrophotometer.
PREPARATION OF SAMPLE
6. See Method 1.
PROCEDURE
Preparation of the standard curve
7.—7.1 Transfer 2, 5, 10, 20, 25 and 50 ml aliquot parts of biuret standard solution (4.6) into a series of six 100 ml graduated flasks. Make up the volumes to about 50 ml with water, add one drop of indicator solution (4.7) and neutralise, if necessary, with 0.1 N sulphuric acid (4.2). Add with swirling 20.0 ml of the alkaline tartrate solution (4.4) and then 20.0 ml copper sulphate solution (4.5). Make up to the mark with water, mix and allow to stand at 30±2°C for fifteen minutes.
At the same time prepare a reagent blank as follows. Place 50 ml water in a 100 ml graduated flask and proceed as described above from “…add one drop of indicator solution….”
Measure the absorbance of each solution at 546 nm against the reagent blank as reference, using cells of a suitable thickness. Plot the calibration curve, using the absorbances as the ordinates and the corresponding quantities of biurent in milligrams, as the abscissae.
Preparation of solution for analysis
7.2 Weigh to the nearest 0.001 g, 10 g of the prepared sample; dissolve in about 150 ml of water in a 250 ml graduated flask and make up to the mark and mix. Filter if necessary.
Note 1 If the sample for analysis contains more than 0.015 g of ammoniacal nitrogen, dissolve it in 50 ml methanol (4.1) in a 250 ml beaker. Reduce by evaporation to a volume of about 25 ml. Transfer quantitatively to a graduated 250 ml flask. Make up to the mark with water. Filter, if necessary, through a dry fluted filter into a dry receiver.
Note 2 Elimination of the opalescence: if any collodial substance is present difficulties may arise during filtering. In that case the solution for analysis is prepared as follows:
dissolve the sample in 150 ml of water, add 2 ml 1 N hydrochloric acid, and filter the solution through two flat very fine filters into a 250 ml graduated flask. Wash the filters with water and make up to volume. Continue the process according to the method described in 7.3.
Determination
7.3 According to the presumed biuret content, transfer with a pipette 25 or 50 ml from the solution prepared in 7.2, to a 100 ml graduated flask and neutralise if necessary with 0.1 N sulphuric acid or sodium hydroxide solution (4.2 or 4.3) as required, using methyl red indicator (4.7). Add 20.0 ml of the alkaline solution of potassium sodium tartrate (4.4) and 20.0 ml of the copper solution (4.5). Make up to volume, mix thoroughly and leave standing for 15 minutes at 30°C±2. Measure the absorbance of the solution as described in 7.1
EXPRESSION OF RESULTS
8.
where
C = weight, in milligrams, of biuret read from the standard curve;
V = volume of the aliquot used for the determination.

where
C = weight, in milligrams, of biuret read from the standard curve;
V = volume of the aliquot used for the determination.
8a.DETERMINATION OF DIFFERENT FORMS OF NITROGEN IN THE SAME SAMPLE—IN THE PRESENCE OF CYANAMIDE NITROGEN
SCOPE
1. This method is for the determination of any one form of nitrogen in the presence of any other form.
FIELD OF APPLICATION
2. Any fertiliser in Group 1(a) of Section A, and Groups 1, 2 and 3 of Section B of the Table in Schedule 1 of the Fertilisers Regulations 1990(14) containing nitrogen in various forms.
PRINCIPLE
Total soluble and insoluble nitrogen
3.—3.1 According to the list of standard fertilisers, this method is applicable to products containing calcium cyanamide.
3.1.1In the absence of fertilisers, the sample is subjected to direct Kjeldahl digestion.
3.1.2In the presence of nitrates, the sample is subjected to Kjeldahl digestion after reduction with the aid of metallic iron and stannous chloride.
In both cases, the ammonia is determined according to Method 2.
Note: If analysis shows an insoluble nitrogen content of more than 0.5%, it is presumed that the fertiliser contains other forms of insoluble nitrogen not specified for fertilisers covered by the list in paragraph 2.
Forms of soluble nitrogen
3.2 The following are determined from different aliquot parts taken from the same solution of the sample:
3.2.1Total soluble nitrogen
3.2.1.1In the absence of nitrates, by direct Kjeldahl digestion.
3.2.1.2In the presence of nitrates, by Kjeldahl digestion on an aliquot part taken from the solution after reduction according to Ulsch, the ammonia being determined in both cases as described in Method 2.
3.2.2Total soluble nitrogen with the exception of nitric nitrogen, by Kjeldahl digestion after elimination in an acid medium of nitric nitrogen with ferrous sulphate, the ammonia being determined as described in Method 2.
3.2.3Nitric nitrogen by difference
3.2.3.1In the absence of calcium cyanamide, between (3.2.1.2) and (3.2.2) or between total soluble nitrogen (3.2.1.2) and the sum of ammoniacal nitrogen and ureic nitrogen (3.2.4 + 3.2.5).
3.2.3.2In the presence of calcium cyanamide, between (3.2.1.2) and (3.2.2) and between (3.2.1.2) and the sum of (3.2.4 + 3.2.5 + 3.2.6).
3.2.4Ammoniacal nitrogen
3.2.4.1Solely in the presence of ammoniacal nitrogen and ammonical + nitric nitrogen, by applying Method 2.
3.2.4.2In the presence of ureic nitrogen and/or cyanamide nitrogen, by cold distillation after making slightly alkaline, the ammonia being absorbed in a standard solution of sulphuric acid and determined as described in Method 2.
3.2.5Urea nitrogen
Either
3.2.5.1.By conversion using urease, into ammonia which is titrated with a standard solution of hydrochloric acid,
or:
3.2.5.2.By gravimetry with xanthydrol, although biuret will also be precipitated by xanthydrol, this should not give rise to a significant error in the determination since its level is generally low in absolute value in compound fertilisers,
or:
3.2.5.3.By difference, according to the following table:
Case Nitric Nitrogen Ammoniacal Nitrogen Cyanamide Nitrogen Difference 1 Absent Present Present (3.2.1.1)-(3.2.4.2 + 3.2.6) 2 Present Present Present (3.2.2)-(3.2.4.2 + 3.2.6) 3 Absent Present Absent (3.2.1.1)-(3.2.4.2) 4 Present Present Absent (3.2.2)-(3.2.4.2)
3.2.6Cyanamide nitrogen, by precipitation as a silver compound, the nitrogen being estimated in the precipitate by the Kjeldahl method.
REAGENTS
4.—4.1 Potassium sulphate.
4.2 Iron power, reduced with hydrogen (the presribed quantity of iron must be able to reduce at least 50 mg of nitric nitrogen).
4.3 Potassium thiocyanate.
4.4 Potassium nitrate.
4.5 Ammonium sulphate.
4.6 Urea.
4.7 Sulphuric acid solution: dilute an appropriate volume of sulphuric acid (d = 1.84 g/ml) with an equal volume of water.
4.8 Sulphuric acid, 0.2 N solution.
4.9 Sodium hydroxide solution, 30 g per 100 ml, ammonia free.
4.10 Sodium or potassium hydroxide, 0.2 N solution, free from carbonates.
4.11 Stannous chloride solution:
dissolve 120 g of stannous chloride (SnCl2.2H2O) in 400 ml of conentrated hydrochloric acid (d = 1.18 g/ml) and make up to 1 litre with water. The solution must be perfectly clear and prepared immediately before use.
It is essential to check the reducing power of stannous chloride: dissolve 0.5 g of stannous chloride in 2 ml of concentrated hydrochloric acid (d = 1.18 g/ml) and make up to 50 ml with water. Then add 5 g of Rochelle salt (potassium sodium tartrate) and a sufficient quantity of sodium bicarbonate for the solution to be alkaline to litmus paper.
Titrate with 0.1 N iodine solution in the presence of a starch solution as an indicator.
1 ml of 0.1 N iodine solution corresponds to 0.01128 g of SnCl2.2H2O.
At least 80% of the total tin present in the solution thus prepared must be in bivalent form. For the titration, at least 35 ml of 0.1 N iodine solution must therefore be used.
4.12 Sulphuric acid, concentrated (d = 1.84 g/ml).
4.13 Hydrochloric acid solution, approximately 30% (W/V) H2SO4.
4.14 Glacial acetic acid.
4.15 Sulphuric acid solution, approximately 30% (W/V) H2SO4.
4.16 Ferrous sulphate, crystalline FeSO4.7H2O.
4.17 Sulphuric acid, 0.1 N solution.
4.18 Octan-1-ol.
4.19 Potassium carbonate, saturated solution.
4.20 Sodium or potassium hydroxide, 0.1 N solution, free from carbonate.
4.21 Barium hydroxide, saturated solution.
4.22 Sodium carbonate solution, 10 g per 100 ml.
4.23 Hydrochloric acid, 2 N solution.
4.24 Hydrochloric acid, 0.1 N solution.
Urease solution:
4.25 suspend 0.5 g of active urease in 100 ml of distilled water.
Using 0.1 N hydrochloric acid (4.24), adjust the pH to 5.4, measured by a pH meter.
4.26 Xanthydrol solution, 5 g per 100 ml in ethanol or methanol (4.31) (do not use products giving a high proportion of insoluble matter). The solution may be kept for three months in a well-stoppered bottle, away from the light.
4.27 Copper oxide (CuO): 0.3 to 0.4 g per determination or an equivalent quantity of copper sulphate pentahydrate of 0.95 to 1.25 g per determination.
4.28 Anti-bump granules washed in hydrochloric acid and ignited.
4.29 Indicator solutions:
4.29.1 Mixed indicator solution:
4.29.1Solution A: dissolve 1 g of methyl red in 37 ml of 0.1 N sodium hydroxide solution and make up to one litre with water.
Solution B: dissolve 1 g of methylene blue in water and make up to one litre.
Mix one volume of solution A and 2 volumes of solution B.
This indicator is violet in acid solution, grey in neutral solution and green in alkaline solution.
Use 0.5 ml (10 drops) of this indicator solution.
4.29.2 Methyl red indicator solution:
4.29.2dissolve 0.1 g of methyl red in 50 ml of 95% ethanol, make up to 100 ml with water and filter if necessary. This indicator (4 to 5 drops) can be used instead of the previous one.
Indicator papers:
4.30 Litmus, bromothymol blue (or other papers sensitive to pH 6 to 8).
4.31 Ethanol or methanol: solution 95%.
APPARATUS
5.—5.1 Distillation apparatus. See Method 2.
5.2 Apparatus for the determination of ammoniacal nitrogen according to analytical technique 7.2.5.3. An example of recommended apparatus is reproduced in Figure 6 in the Appendix.
The apparatus is made up of a specially shaped receptacle with a ground glass neck, a side neck, a connecting tube with a splash head and a perpendicular tube for the introduction of air. The tubes can be connected to the receptacle by means of a simple perforated rubber bung. It is important to give a suitable shape to the end of the tubes introducing air, since the bubbles of gas must be evenly distributed throughout the solutions contained in the receptacle and the absorber. The best arrangement consists of small mushroom-shaped pieces with an external diameter of 20 mm and six openings of 1 mm around the periphery.
5.3 Apparatus for the estimation of urea nitrogen according to the urease technique (7.2.6.1).
It consists of a 300 ml Erlenmeyer flask, with a seperating funnel and a small absorber. An example of recommended apparatus is reproduced in Figure 7 in the Appendix.
5.4 Rotary shaker, 35-40 turns per minute.
5.5 pH meter.
5.6 Laboratory oven.
5.7 Sintered glass crucibles, diameter of pores 5 to 15 microns.
PREPARATION OF THE SAMPLE
6. See Method 1.
PROCEDURE
7.—7.1 Total soluble and insoluble nitrogen
7.1.1In the absence of nitrate
7.1.1.1Digestion
Weigh to the nearest 0.001 g, a quantity of the prepared sample containing 100 mg of nitrogen at the most. Place it in the flask of the distillation apparatus (5. 1). Add 10 to 1 5 g of potassium sulphate (4. 1), the prescribed quantity of catalyst (4.27), and a few anti-bump granules (4.28). Then add 50 ml of dilute sulphuric acid (4.7), and mix thoroughly. First heat gently, mixing from time to time, until foaming ceases. Then heat so that the liquid boils steadily and keep it boiling for one hour after the solution has become clear, preventing any organic matter from sticking to the sides of the flask. Allow to cool. Carefully add about 350 ml of water, with mixing. Ensure than the dissolution is as complete as possible. Allow to cool and connect the flask to the distillation apparatus (5.1).
7.1.1.2Distillation of ammonia
Transfer with a pipette, into the receiver of the apparatus, 50 ml standard 0.2 N sulphuric acid (4.8). Add the indicator (4.29.1 or 4.29.2). Ensure that the tip of the condenser is at least 1 cm below the level of the solution.
Taking the necessary precautions to avoid any loss of ammonia, carefully add to the distillation flask enough of the concentrated sodium hydroxide solution (4.9) to make the liquid stongly alkaline (120 ml is generally sufficient: check by adding a few drops of phenolphthalein. At the end of the distillation the solution in the flask must still be clearly alkaline). Adjust the heating of the flask so as to distil 150 ml in half an hour. Test with indicator paper (4.30) that the distillation has been completed. If it has not, distil a further 50 ml and repeat the test until the supplementary distillate reacts neutrally to the indicator paper (4.30). Then lower the receiver, distil a few ml more and rinse the tip of the condenser. Titrate the excess acid with a standard solution of potassium or sodium hydroxide 0.2 N (4.10) to the end point of the indicator.
7.1.1.3Blank test
Make a blank test under the same conditions (omitting only the sample) and use this value in the calculation of the final result.
7.1.1.4Expression of the result
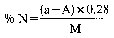
where:
a= ml of standard solution of sodium or potassium hydroxide (0.2 N) used for the blank, carried out by placing in the receiver of the apparatus (5.1), 50.0 ml of standard solution of sulphuric acid (0.2 N) (4.8).
A= ml of standard solution of sodium or potassium hydroxide (0.2 N) used for the analysis.
M=weight of the sample in grams.
7.1.2In the presence of nitrate
7.1.2.1Test sample
Weigh to the nearest 0.001 g, a quantity of the sample containing not more than 40 mg of nitric nitrogen.
7.1.2.2Reduction of the nitrate
Mix the sample in a small mortar with 50 ml of water. Transfer with the minimum amount of distilled water into a 500 ml Kjeldahl flask. Add 5 g of reduced iron (4.2) and 50 ml of stannous chloride solution (4.11). Shake and leave it to stand for half an hour. During this time shake again after 10 and 20 minutes.
7.1.2.3Kjeldahl digestion
Add 30 ml of sulphuric acid (4.12), 5 g of potassium sulphate (4.1), the prescribed quantity of catalyst (4.27) and some anti-bump granules (4.28). Heat gently with the flask slightly tilted. Increase the heat slowly and shake the solution frequently to keep the mixture suspended; the liquid darkens and then clears with the formation of a yellow-green anhydrous iron sulphate suspension. Continue heating for one hour after obtaining a clear solution, maintaining it at simmering point. Leave to cool. Cautiously take up the contents of the flask in a little water and add little by little 100 ml of water. Mix and transfer the contents of the flask into a 500 ml graduated flask. Rinse the flask several times with distilled water. Make up the volume with water and mix. Filter through a dry filter into a dry receiver.
7.1.2.4Distillation of ammonia
Transfer by pipette, into the flask of the distillation apparatus (5.1), an aliquot part containing 100 mg of nitrogen at the most. Dilute to about 350 ml with distilled water, add a few anti-bump granules (4.28), connect the flask to the distillation apparatus and continue the estimation as described in paragraph 7.1.1.2.
7.1.2.5Blank test
See 7.1.1.3.
7.1.2.6Expression of the result
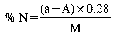
where:
a = ml of standard solution of sodium or potassium hydroxide (0.2 N) used for the blank, carried out by placing in the receiver of the apparatus (5.1), 50.0 ml of standard solution of sulphuric acid (0.2 N) (4.8).
A = ml of standard solution of sodium or potassium hydroxide (0.2N) used for the analysis.
M = weight of the sample, expressed in grams, present in the aliquot part taken for analysis.
7.2 Forms of soluble nitrogen
7.2.1 Preparation of the solution to be analysed
7.2.1Weigh to the nearest 0.001 g, 10 g of the sample and place it in a 500 ml graduated flask,
7.2.1.1In the case of fertilisers not containing cyanamide nitrogen
Add to the flask 50 ml of water and then 20 ml of dilute hydrochloric acid (4.13). Shake and leave it to stand until the evolution of carbon dioxide ceases. Then add 400 ml of water and shake for half an hour on the rotary shaker (5.4).
Make up to the volume with water, mix and filter through a dry filter into a dry receiver.
7.2.1.2In the case of fertilisers containing cyanamide nitrogen
Add to the flask 400 ml of water and a few drops of methyl red (4.29.2). If necessary make the solution acid by using acetic acid (4.14). Add 15 ml of acetic acid (4.14). Shake on the rotary shaker (5.4) for 2 hours. If necessary, re-acidify the solution during the operation, using acetic acid (4.14). Make up to the volume with water, mix, filter immediately through a dry filter into a dry receiver and immediately determine the cyanamide nitrogen.
In both cases, determine the various soluble forms of nitrogen the same day the solution is made up, starting with the cyanamide nitrogen and urea nitrogen, if they are present.
7.2.2Total soluble nitrogen
7.2.2.1In the absence of nitrate
Transfer by pipette into a 300 ml Kjeldahl flask, an aliquot part of the filtrate (7.2.1.1 or 7.2.1.2), containing 100 mg of nitrogen at the most. Add 15 ml of concentrated sulphuric acid (4.12), 0.4 g of copper oxide or 1.25 g of copper sulphate (4.27) and a few anti-bump granules (4.28). First heat gently to begin the digestion and then at a higher temperature until the liquid becomes colourless or slightly greenish and white fumes are clearly apparent. After cooling, quantitatively transfer the solution into the distillation flask, dilute to about 500 ml with water and add a few anti-bump granules (4.28). Connect the flask to the distillaton apparatus (5. 1) and continue the distillation as determination in paragraph 7.1.1.2.
7.2.2.2In the Presence of nitrate
Transfer by pipette into a 500 ml Erlenmeyer flask, an aliquot part of the filtrate (7.2.1.1 or 7.2.1.2) containing not more than 40 mg of nitric nitrogen. At this stage of the analysis the total quantity of nitrogen is not important. Add 10 ml of 30% sulphuric acid (4.15), 5 g of reduced iron (4.2) and immediately cover the Erlenmeyer flask with a watch glass. Heat gently until the reaction is steady but not vigorous. At this juncture stop the heating and allow the flask to stand for at least three hours at ambient temperature. With water, quantitatively transfer the liquid into a 250 ml graduated flask, leaving behind the undissolved iron and make up to the mark with water. Mix thoroughly, and transfer by pipette into a 300 ml Kjeldahl flask, an aliquot part containing 100 mg of nitrogen at the most. Add 15 ml of concentrated sulphuric acid (4.12), 0.4 g of copper oxide or 1.25 g of copper sulphate (4.27) and some anti-bump granules (4.28). First heat gently to begin the digestion and then at a higher temperature until the liquid becomes colourless or slightly greenish and white fumes are clearly apparent. After cooling, quantitatively transfer the solution into the distillation flask, dilute to approximately 500 ml with water and add some anti-bump granules (4.28). Connect the flask to the distillation apparatus (5.1) and continue the determination as described in paragraph 7.1.1.2.
7.2.2.3Blank test
See 7.1.1.3.
7.2.2.4Expression of the result

where:
a= ml of standard solution of sodium or potassium hydroxide (0.2 N) used for the blank, carried out by placing in the receiver of the apparatus (5.1), 50.0 ml of standard solution of sulphuric acid (0.2 N) (4.8).
A = ml of standard solution of sodium or potassium hydroxide (0. 2 N) used for analysis.
M=weight of the sample, expressed in grams, present in the aliquot part taken for analysis.
7.2.3 Total soluble nitrogen with the exception of nitric nitrogen
7.2.3Transfer by pipette into a 300 mi Kjeldahl flask, an aliquot part of the filtrate (7.2.1.1 or 7.2.1.2) containing not more than 50 mg of nitrogen to be determined. Dilute to 100 ml with water, add 5 g of ferrous sulphate (4.16), 20 ml of concentrated sulphuric acid (4.1) and some anti-bump granules (4.28). First heat gently and then increase the heat until white fumes appear. Continue the digestion for 15 minutes. Stop the heating, introduce the copper oxide (4.27) as a catalyst and keep it at a temperature such that white fumes are emitted for a further 10 to 15 minutes. After cooling, quantitatively transfer the contents of the Kjeldahl flask into the distillation flask of the apparatus (5. 1). Dilute to approximately 500 ml with water and add a few anti-bump granules (4.28). Connect the flask to the distillaton apparatus and continue the determinaton as described in paragraph 7.1.1.2.
7.2.3.1Blank test
See 7.1.1.3.
7.2.3.2Expression of result
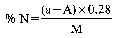
where:
a = ml of standard solution of sodium or potassium hydroxide (0.2 N) used for the blank, carried out by placing in the receiver of the apparatus (5.1), 50 ml of the standard sulphuric acid solution (0.2N) (4.8).
A = ml of standard solution of sodium or potassium hydroxide (0.2 N) used for the analysis.
M = weight of the sample, expressed in grams, present in the aliquot part taken for analysis.
7.2.4Nitric nitrogen is obtained:
7.2.4.1In the absence of calcium cyanamide
By the difference between the results obtained in paragraphs 7.2.2.4 and 7.2.3.2 and/or the result obtained in paragraph 7.2.2.4 and the sum of the results obtained in paragraphs 7.2.5.2 or 7.2.5.5 and 7.2.6.3 or 7.2.6.5 or 7.2.6.6.
7.2.4.2In the presence of calcium cyanamide
By the difference between the results obtained in paragraphs 7.2.2.4 and 7.2.3.2 and between the result obtained in paragraph 7.2.2.4 and the sum of the results obtained in paragraphs 7.2.5.5 and 7.2.6.3 or 7.2.6.5 or 7.2.6.6 and 7.2.7.
7.2.5Ammoniacal nitrogen
7.2.5.1Solely in the presence of ammoniacal nitrogen and ammoniacal+nitric nitrogen
Transfer by pipette into the flask of the distillation apparatus (5.1) an aliquot part of the filtrate (7.2.1.1) containing 100 mg of ammoniacal nitrogen at the most. Add water to obtain a total volume of about 350 ml and some anti-bump granules (4.28) to facilitate boiling. Connect the flask to the distillation apparatus, add 20 ml of sodium hydroxide solution (4.9) and distil as described in paragraph 7.1.1.2.
7.2.5.2Expression of result
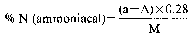
where:
a = ml of standard solution of sodium or potassium hydroxide (0.2 N) used for the blank, carried out by placing in the receiver of the apparatus (5.1), 50 ml of the standard sulphuric acid solution (0.2 N) (4.8).
A = ml of standard solution of sodium or potassium hydroxide (0.2 N) used for the analysis.
M = weight of the sample, expressed in grams, present in the aliquot part taken for analysis.
7.2.5.3In the presence of urea and/or cyanamide nitrogen
Transfer by pipette into the dry flask of the apparatus (5.2), an aliquot part of the filtrate (7.2.1.1 or 7.2.1.2) containing 20 mg of ammoniacal nitrogen at the most. Then assemble the apparatus. Transfer by pipette into the 300 ml Erlenmeyer flask 50 ml of the standard sulphuric acid solution 0.1 N (4.17) and enough distilled water for the level of the liquid to be approximately 5 cm above the opening of the delivery tube; add the indicator (4.29.1). Introduce, through the side neck of the reaction flask, distilled water to make up the volume to about 50 ml and mix. To avoid foaming during aeration, add a few drops of octan-1-o1 (4.18). Make the solution alkaline by adding 50 ml of saturated potassium carbonate solution (4.19) and immediately begin to expel the ammonia thus liberated from the cold suspension. A strong current of air is necessary (flow of approximately 3 litres per minute) and should be purified beforehand by passing it through washing flasks containing dilute sulphuric acid and dilute sodium hydroxide. Instead of using pressurised air, it is also possible to use a vacuum (water pump) provided that the inflow tube is connected in a sufficiently airtight manner to the receiver used to collect the ammonia. The liberation of the ammonia is generally complete after three hours. It is nevertheless advisable to verify this by changing the receiving flask. When the operation is finished, disconnect the flask from the apparatus, rinse the tip of the tube and the sides of the flask with a little distilled water. Titrate the excess acid with standard sodium hydroxide solution (0.1 N) (4.20) to the end point of the indicator (4.29.1).
7.2.5.4Blank test
See 7.1.1.3.
7.2.5.5Expression of the result
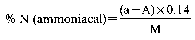
where:
a = ml of standard solution of sodium or potassium hydroxide (0.1 N) used for the blank, carried out by placing in the 300 ml Erlenmeyer flask of the apparatus (5.2), 50 ml of the standard solution of sulphuric acid (0.1 N) (4.17).
A = ml of standard solution of sodium or potassium hydroxide (0.1 N) used for the analysis.
M = weight of the sample, expressed in grams, present in the aliquot part taken for analysis.
7.2.6Urea nitrogen
7.2.6.1Urease method
Transfer by pipette into a 500 ml graduated flask, an aliquot part of the filtrate (7.2.1.1 or 7.2.1.2) containing not more than 250 mg of urea nitrogen. To remove phosphates add saturated barium hydroxide solution (4.21) until no further precipitaton occurs. Eliminate the excess of barium ions and any dissolved calcium ions by adding 10% sodium carbonate solution (4.22). Allow the precipitate to settle and check whether total Precipitation has occurred. Make up to the mark, mix and filter through a pleated filter. Transfer by pipette 50 ml of the filtrate into the 300 ml Erlenmeyer flask of the apparatus (5.3). Acidify the filtrate with 2 N hydrochloric acid (4.23), until pH of 3.0 measured by the pH meter (5.5) is obtained. Then raise the pH to 5.4 with 0.1 N sodium hydroxide (4.20).
To avoid losses of ammonia during decomposition by the urease, close the Erlenmeyer flask with a stopper provided with a separating funnel and a small bubble trap containing exactly 2 ml of standard 0.1 N hydrochloric acid (4.24). Introduce through the separating funnel 20 ml of urease solution (4.25), and allow to stand for one hour at 20-25°c.Transfer by pipette 25 ml of standard 0.1 N hydrochloric acid (4.24) into the separating funnel, allow it to run through into the solution and then rinse with a little water. In the same way quantitatively transfer the contents of the bubble trap into the solution contained in the Erlenmeyer flask. Titrate the excess acid with the standard solution of sodium hydroxide (0.1 N) (4.20), until a pH of 5.4 is obtained, measured by the pH meter.
7.2.6.2Blank test
See 7.1.1.3.
7.2.6.3Expression of result

where:
a = ml of standard solution of sodium of potassium hydroxide (0.1 N) used for the blank, carried out exactly under the same conditions as the analysis.
A = ml of standard solution of sodium of potassium hydroxide (0. I N) used for the analysis.
M = weight of the sample, expressed in grams, present in the aliquot part taken for analysis.
(1)Remarks
After precipitation by the solutions or barium hydroxide and sodium carbonate, make up to the mark, filter and neutralise as rapidly as possible.
(2)The titration may also be carried out with the indicator (4.29.2), but the end point is then more difficult to observe.
7.2.6.4Gravimetric method with xanthydrol
Transfer by pipette into a 250 ml beaker, an aliquot part of the filtrate (7.2.1.1 or 7.2.1.2) containing not more than 20 mg of urea. Add 40 ml of acetic acid (4.14). Stir with a glass rod for one minute, allow any precipitate to settle for five minutes. Filter into a 100 ml beaker, wash with several ml of acetic acid (4.14), then add to the filtrate drop by drop, 10 ml of xanthydrol solution (4.26), stirring continuously with a glass rod. Allow to stand until the precipitate appears, then stir again for one or two minutes. Allow to stand for one and a half hours. Filter through a sintered glass crucible (5.7) which has been previously dried and weighed, using a slight reduction in pressure. Wash three times with 5 ml ethanol (4.31) without trying to remove all the acetic acid. Place it in the oven (5.6) at a temperature of 130°C for one hour (do not exceed 145°C). Allow to cool in a desiccator and weigh.
7.2.6.5Expression of result

where:
m=weight of the precipitate obtained, in grams.
M=weight of the sample, in grams, present in the aliquot part taken for analysis. Correct for the blank.
Note: although biuret will also be precipitated by xanthydrol, this should not give rise to a significant error in the determination since its level is generally low.
7.2.6.6Method by difference
Urea nitrogen may also be calculated according to the following table:—
| Case | Nitric Nitrogen | Ammoniacal Nitrogen | Cyanamide Nitrogen | Ureic Nitrogen |
|---|---|---|---|---|
| 1 | Absent | Present | Present | (7.2.2.4)-(7.2.5.5+7.2.7) |
| 2 | Present | Present | Present | (7.2.3.2)-(7.2.5.5+7.2.7) |
| 3 | Absent | Present | Absent | (7.2.2.4)-(7.2.5.5) |
| 4 | Present | Present | Absent | (7.2.3.2)-(7.2.5.5) |
7.2.7 Cyanamide Nitrogen
7.2.7Take an aliquot part of the filtrate (7.2.1.2), containing 10 to 30 mg of cyanamide nitrogen and place it in a 250 ml beaker. Continue the analysis according to Method 6.
VERIFICATION OF THE RESULTS
8.—8.1 In certain cases, a difference may be found between the total nitrogen obtained directly from a weighed out sample (paragraph 7.1) and total soluble nitrogen (paragraph 7.2.2). Nevertheless, the difference should not be greater than 0.5%. If this is not the case, the fertiliser contains forms of insoluble nitrogen not specified for fertilisers covered by the list in paragraph 2.
8.2 Before each analysis, check that the apparatus is working properly and that the correct application of the method is used, with a standard solution including the various forms of nitrogen in proportions similar to those of the test sample. This standard solution is prepared from solutions of potassium thiocyanate (4.3), potassium nitrate (4.4), ammonium sulphate (4.5) and urea (4.6).
8b.DETERMINATION OF DIFFERENT FORMS OF NITROGEN IN THE SAME SAMPLE—IN THE ABSENCE OF CYANAMIDE NITROGEN
SCOPE
1. This method is for the determination of any one form of nitrogen in the presence of any other form, in the absence of cyanamide nitrogen.
FIELD OF APPLICATION
2. This method is applicable to all fertilisers in Group 1(a) of Section A and Groups 1, 2 and 3 of Section B of the Table in Schedule 1 of the Fertilisers Regulations 1990(15) which contain exclusively nitric, ammoniacal or ureic nitrogen.
PRINCIPLE
3. The following determinations are made on different portions of a single sample solution.
3.1 Total soluble nitrogen
3.1.1In the absence of nitrates, by direct Kjeldahl digestion of the solution.
3.1.2In the presence of nitrates, by Kjeldahl digestion of a portion of the solution after reduction by the Ulsch method; ammonia is determined in both cases as described in Method 2.
3.2 Total soluble nitrogen except nitric nitrogen, by Kjeldahl digestion after elimination of nitric nitrogen in acid medium by means of ferrous sulphate; ammonia is determined as described in Method 2.
3.3 Nitric nitrogen, by difference: between 3.1.2 and 3.2 and/or between total soluble nitrogen (3.1.2) and the sum of ammoniacal and ureic nitrogen (3.4 + 3.5).
3.4 Ammoniacal nitrogen, by cold distillation of a weak alkaline solution; the ammonia is obtained in a solution of sulphuric acid and determined as described in Method 2.
3.5 Ureic nitrogen, either:
3.5.1By conversion using urease, into ammonia, which is determined by titration with a standard solution of hydrochloric acid,
or:
3.5.2By gravimetry using xanthydrol: although biuret will also be precipitated by xanthydrol, this should not give rise to a significant error in the determination since its level is generally low in absolute value in compound fertilisers,
or:
3.5.3By difference, according to the following table:
| Case | Nitric nitrogen | Ammoniacal nitrogen | Difference |
|---|---|---|---|
| 1 | Absent | Present | (3.1.1)-(3.4) |
| 2 | Present | Present | (3.2)-(3.4) |
REAGENTS
4.—4.1 Potassium sulphate.
4.2 Iron powder, reduced with hydrogen (the prescribed quantity of iron must be able to reduce at least 50 mg nitric nitrogen).
4.3 Potassium nitrate.
4.4 Ammonium sulphate.
4.5 Urea.
4.6 Sulphuric acid, 0.2 N solution.
4.7 Sodium hydroxide solution, 30 g per 100 ml, ammonia free.
4.8 Sodium or potassium hydroxide, 0.2 N solution, free of carbonates.
4.9 Sulphuric acid (d = 1.84 g/ml).
4.10 Hydrochloric acid solution: dilute an appropriate volume of hydrochloric acid (d = 1.18 g/ml) with an equal volume of water.
4.11 Glacial acetic acid.
4.12 Sulphuric acid, solution approximately 30% (W/V) H2SO4.
4.13 Ferrous sulphate, crystalline FeSO4.7H2O.
4.14 Sulphuric acid, 0.1 N solution.
4.15 Octan-1-ol.
4.16 Potassium carbonate, saturated solution.
4.17 Sodium or potassium hydroxide, 0.1 N solution.
4.18 Barium hydroxide, saturated solution.
4.19 Sodium carbonate solution, 10 g per 100 ml.
4.20 Hydrochloric acid, 2 N solution.
4.21 Hydrochloric acid, 0.1 N solution.
4.22 Urease solution: suspend 0.5 g active urease in 100 ml distilled water. Using 0.1 N hydrochloric acid (4.21), adjust pH to 5.4, measured with pH meter.
4.23 Xanthydrol solution, 5 g per 100 ml in ethanol or methanol (4.28) (do not use products giving a high proportion of insoluble material). The solution can be kept for 3 months in a carefully stoppered bottle in darkness.
4.24 Catalyst: copper oxide (CuO), 0.3 to 0.4 g per determination, or an equivalent amount of copper sulphate pentahydrate, 0.95 to 1.25 g per determination.
4.25 Anti-bump granules of pumice stone washed with hydrochloric acid and ignited.
4.26 Indicator solutions:
4.26.1 Mixed indicator:
4.26.1Solution A: dissolve 1 g methyl red in 37 ml 0.1 N sodium hydroxide solution and make up to 1 litre with water.
Solution B: dissolve 1 g methylene blue in water and make up to 1 litre.
Mix 1 volume of solution A and 2 volumes of solution B. This indicator is violet in acid solution, grey in neutral solution and green in alkaline solution: use 0.5 ml (10 drops) of this indicator.
4.26.2 Methyl red indicator solution:
4.26.2dissolve 0.1 g methyl red in 50 ml 95% ethanol, make up to 100 ml with water and filter if necessary; 4-5 drops of this indicator can be used instead of the previous one.
4.27 Indicator papers: litmus, bromothymol blue (or other papers sensitive to pH 6-8).
4.28 Ethanol or methanol, 95% (V/V).
APPARATUS
5.—5.1 Distillation apparatus. See Method 2.
5.2 Apparatus for determination of ammoniacal nitrogen. An example of recommended apparatus is reproduced in Figure 6 in the Appendix.
5.3 Apparatus for determination of urea nitrogen by the urease method (7.6.1). An example of recommended apparatus is reproduced in Figure 7 in the Appendix.
5.4 Rotary shaker: 35-40 turns per minute.
5.5 pH meter.
5.6 Sintered glass crucibles, diameter of pores 5 to 15 microns.
PREPARATION OF SAMPLE
6. See Method 1.
PROCEDURE
Preparation of solution for analysis
7.—7.1 Weigh to the nearest 0.001 g, 10 g of the prepared sample and transfer to a 500 ml graduated flask. Add 50 ml water and then 20 ml dilute hydrochloric acid (4.10) and shake. Allow to stand until the evolution of carbon dioxide ceases. Add 400 ml water; shake for half an hour; make up to volume with water, mix, filter on a dry filter into a dry container.
7.2 Total nitrogen
7.2.1 In the absence of nitrates
7.2.1Transfer by pipette into a 300 ml Kjeldahl flask a portion of the filtrate (7.1), containing a maximum of 100 mg nitrogen. Add 15 ml concentrated sulphuric acid (4.9), 0.4 g copper oxide or 1.25 g copper sulphate (4.24) and a few glass beads to control boiling. Heat moderately at first in order to initiate the reaction, then more strongly until the liquid becomes colourless or slightly greenish and white fumes appear. After cooling, transfer the solution into the distillation flask, dilute to about 500 ml with water and add a few granules of pumice stone (4.25). Connect the flask to the distillation apparatus (5.1) and carry out the determination as described in Method 8a, 7.1.12.
7.2.2 In the presence of nitrates
7.2.2Transfer by pipette into a 500 ml Erlenmeyer flask a portion of the filtrate (7.1) containing not more than 40 mg nitric nitrogen. At this this stage of the analysis, the total quantity of nitrogen is unimportant. Add 10 ml 30% sulphuric acid (4.12), 5 g reduced iron (4.2) and immediately cover the Erlenmeyer flask with a watch glass. Heat gently until the reaction becomes strong but not violent. Stop heating and allow to stand for at leqast 3 hours at ambient temperature. Transfer the liquid quantitatively to a 250 nml graduated flask, ignoring undissolved iron. Make up to the mark with water and mix carefully. Transfer by pipette a portion containing a maximum of 100 mg nitrogen into a 300 ml Kjeldahl flask. Add 15 ml concentrated sulphuric acid (4.9), 0.4 g copper oxide or 1.25 g copper sulphate (4.24) and a few glass beads.
Heat moderately at first in order to initiate the reaction, then more strongly until the liquid becomes colourless or slightly greenish and white fumes appear. After cooling, transfer the solution quantitatively to the distillation flask, dilute to about 500 ml with water and add a few granules of pumice stone (4.25). Connect the flask to the distillation apparatus (5.1) and continue the determination as described in Method 8a, 7.1.1.2.
7.2.3 Blank test
7.2.3Make a blank test in the same conditions (omitting only the sample), and use this value in the calculation of the final result.
7.2.4 Expression of result
7.2.4 where:
a = ml of standard solution of sodium or potassium hydroxide (0.2 N) used for the blank, carried out under the same conditions as the analysis.
A = ml of standard solution of sodium or potassium hydroxide (0.2 N) used for the analysis.
M = weight of the sample, in grams, present in the aliquot part taken for analysis.

where:
a = ml of standard solution of sodium or potassium hydroxide (0.2 N) used for the blank, carried out under the same conditions as the analysis.
A = ml of standard solution of sodium or potassium hydroxide (0.2 N) used for the analysis.
M = weight of the sample, in grams, present in the aliquot part taken for analysis.
Total nitrogen excluding nitric nitrogen
7.3 7.3.1 Transfer by pipette into a 300 ml Kjeldahl flask an aliquot part of filtrate (7.1) containing not more than 50 mg of nitrogen to be determined. Dilute to 100 ml with water, add 5 g ferrous sulphate (4.13), 20 ml concentrated sulphuric acid (4.9) and a few glass beads to control boiling (4.29). Heat moderately at first then more strongly until white fumes appear. Continue the reaction for 15 minutes. Stop heating, introduce 0.4 g copper oxide or 1.25 g copper sulphate (4.24) as catalyst, resume heating and maintain production of white fumes for 10-15 minutes. After cooling, transfer the contents of the Kjeldahl flask quantitatively to the distillation flask (5.1). Dilute to about 500 ml with water and add a few granules of pumice stone (4.2). Connect the flask to the distillation apparatus and continue the determination as in Method 8a, 7.1.1.2.
7.3.2 Blank test
7.3.2See 7.2.3.
7.3.3. Expression of result
7.3.3. where:
a = ml of standard solution of sodium or potassium hydroxide (0.2 N) used for the blank, carried out by placing in the receiver of the apparatus (5.1), 50.0 ml of standard solution of sulphuric acid (0.2 N) (4.6).
A = ml of standard solution of sodium or potassium hydroxide (0.2 N) used for the analysis.
M = weight of the sample, in grams, present in the aliquot part taken for analysis.

where:
a = ml of standard solution of sodium or potassium hydroxide (0.2 N) used for the blank, carried out by placing in the receiver of the apparatus (5.1), 50.0 ml of standard solution of sulphuric acid (0.2 N) (4.6).
A = ml of standard solution of sodium or potassium hydroxide (0.2 N) used for the analysis.
M = weight of the sample, in grams, present in the aliquot part taken for analysis.
Nitric nitrogen is obtained: by difference between
7.4 (7.2.4)-(7.5.3 + 7.6.3)
or (7.2.4)-(7.5.3 + 7.6.5)
or (7.2.4)-(7.5.3 + 7.6.6)
Ammoniacal nitrogen
7.5.1 In the presence of ureic nitrogen
7.5.1Transfer by pipette into the dry flask of the apparatus (5.2) a portion of filtrate (7.1) containing a maximum of 20 mg ammoniacal nitrogen. Connect up the apparatus. Place in the 300 ml Erlenmeyer flask 50.0 ml standard 0.1 N sulphuric acid solution (4.14) and an amount of distilled water such that the level of the liquid is about 5 cm above the opening of the intake tube. Introduce through the side neck of the reaction flask distilled water so as to bring the volume to about 50 ml. Shake. To avoid foaming during aeration add several drops of octan-1-ol (4.15). Add 50 ml saturated potassium carbonate solution (4.16) and immediately begin to expel the ammonia thus released from the cold suspension. A strong current of air is necessary (flow rate of about 3 litres per minute) and should be purified by passing ot through washing flasks containing dilute sulphuric acid and dilute sodium hydroxide. Instead of using air under pressure, a vacuum may be used (wtaer pump) provided that the connections between the apparatus are air tight. The liberration of ammonia is generally complete after three hours. However, it is desirable to make certain of this by changing the Erlenmeyer flask. When the process is finished, disconnect the Erlenmeyer flask from the apparatus, rinse the end of the intake tube and the walls of the Erlenmeyer flask with a little distilled water and titrate the excess acid against standard 0.1 N sodium hydroxide solution (4.17).
7.5.2 Blank test
7.5.2See 7.2.3.
7.5.3 Expression of result
7.5.3 where:
a = ml of standard solution of sodium or potassium hydroxide (0.1 N) (4.17) used for the blank, carried out by placing in the receiver of the apparatus (5.2), 50.0 ml of standard solution of sulphuric acid (0.1 N) (4.14).
A = ml of standard solution of sodium or potassium hydroxide (0.1 N) (4.17) used for the analysis.
M = weight of the sample, in grams, present in the aliquot part taken for analysis.

where:
a = ml of standard solution of sodium or potassium hydroxide (0.1 N) (4.17) used for the blank, carried out by placing in the receiver of the apparatus (5.2), 50.0 ml of standard solution of sulphuric acid (0.1 N) (4.14).
A = ml of standard solution of sodium or potassium hydroxide (0.1 N) (4.17) used for the analysis.
M = weight of the sample, in grams, present in the aliquot part taken for analysis.
Ureic acid
7.6.1 Urease method
7.6.1Transfer by pipette into a 500 ml graduated flask, a portion of filtrate (7.1) containing not more than 250 mg of ureic nitrogen. To remove phosphates, add a suitable quantity of saturated barium hydroxide solution (4.18) until further addition does not cause the production of more precipitate. Eliminate excess barium ions (and any dissolved calcium ions) by means of 10% sodium carbonate solution (4.19). Allow to settle and check whether precipitation is complete. Make up to the mark, mix and filter through a fluted filter. Transfer by pipette 50 ml filtrate into the 300 ml Erlenmeyer Flask of the apparatus (5.3). Acidify with 2 N hydrochloric acid (4.20) to pH 3.0, measured by means of the pH meter (5.5). Raise the pH to 5.4 by the addition of 0.1 N sodium hydroxide (4.17). To avoid ammonia losses when hydrolysis by urease occurs, close the Erlenmeyer flask by means of a stopper provided with a dropping funnel and a small bubble trap containing exactly 2 ml standard 0.1 N hydrochloric acid solution (4.21). Intorduce through the seperating funnel, 20 ml urease solution (4.22). Allow to stand for one hour at 20-25°C. Place 25.0 ml of the standard 0.1 N hydrochloric acid solution (4.2) in the dropping funnel, allow to run into the solution, then rinse with a little water. Transfer quantitatively the contents of the bubble trap to the solution contained in the Erlenmeyer flask. Titrate the excess acid using the standard 0.1 N sodium hydroxide solution (4.17), until a pH of 5.4 is obtained, measured on the pH meter.
Remarks
1. After precipitation by the barium hydroxide and sodium carbonate solutions, make up to the mark, filter and neutralise as quickly as possible.
2. The titration may also be carried out using an indicator (4.26), although the change of colour is more difficult to observe.
7.6.2 Blank test
See 7.2.3
7.6.3 Expression of result
where:
a = ml of standard solution of sodium or potassium hydroxide (0.1 N) (4.17) used for the blank, carried out in exactly the same conditions as the analysis.
A = ml of standard solution of sodium or potassium hydroxide (0.1 N) (4.17) used for the analysis.
M = weight of the sample, in grams, present in the aliquot part taken for analysis.

where:
a = ml of standard solution of sodium or potassium hydroxide (0.1 N) (4.17) used for the blank, carried out in exactly the same conditions as the analysis.
A = ml of standard solution of sodium or potassium hydroxide (0.1 N) (4.17) used for the analysis.
M = weight of the sample, in grams, present in the aliquot part taken for analysis.
7.6.4 Gravimetric method using xanthydrol
Transfer by pipette into a 100 ml beaker a portion of the filtrate (7.1) containing not more than 20 mg urea. Add 40 ml acetic acid (4.11). Stir with a glass rod for one minute. Allow any precipitate to settle for five minutes. Filter, wash with a few ml acetic acid (4.11). Add 10 ml xanthydrol solution (4.23) to the filtrate drop by drop, stirring continuously with a glass rod. Allow to stand until the precipitate appears, then stir again for one or two minutes. Allow to stand for one and half hours. Filter through a sintered glass crucible (5.6) which has been previously dried and weighed, using a slight reduction in pressure. Wash three times with 5 ml ethanol (4.28), without trying to remove all the acetic acid. Place in an oven at a temperature of 130°C for one hour (do not exceed 145°C). Allow to cool in a desiccator and weigh.
7.6.5 Expression of result
where:
m = weight of the precipitate in grams.
M = weight of the sample, in grams, present in the aliquot part taken for analysis.
Correct for the blank.
Note: Although biuret will also be precipitated by xanthydrol, this should not give rise to a significant error in the determination since its level is generally low in absolute value in compound fertilisers.

where:
m = weight of the precipitate in grams.
M = weight of the sample, in grams, present in the aliquot part taken for analysis.
Correct for the blank.
Note: Although biuret will also be precipitated by xanthydrol, this should not give rise to a significant error in the determination since its level is generally low in absolute value in compound fertilisers.
7.6.6 Difference method
Ureic N can also be calculated as indicated in the following table:
| Case | Nitric N | Ammoniacal N | Ureic N |
|---|---|---|---|
| 1 | Absent | Present | (7.2.4)-(7.5.3) |
| 2 | Present | Present | (7.3.3)-(7.5.3) |
EXPRESSION OF RESULTS
8.—8.1 Before each analysis, check the functioning of the apparatus and the correct application of the methods used with a standard solution containing the different forms of nitrogen in proportions similar to those in the sample. This standard solution is prepared from solutions of potassium nitrate (4.3), ammonium sulphate (4.4) and urea (4.5).
9a.EXTRACTION OF TOTAL PHOSPHORUS BY MINERAL ACIDS
SCOPE
1. This method is for the determination of phosphorus soluble in mineral acids.
FIELD OF APPLICATION
2. Applicable exclusively to the phosphate fertilisers listed in Group 2(a) of section A and Groups 1, 2 and 4 of Section B of the Table in Schedule 1 of the Fertilisers Regulations 1990(16).
PRINCIPLE
3. Extraction of the phosphorus in the fertiliser with a mixture of nitric acid and sulphuric acid.
REAGENTS
4.—4.1 Sulphuric acid (d = 1.84 g/ml).
4.2 Nitric acid (d = 1.40 g/ml).
APPARATUS
5.—5.1 A Kjeldahl flask, with a capacity of at least 500 ml, or a 250 ml round-bottomed flask with a glass tube forming a reflux condenser.
PREPARATION OF THE SAMPLE
6. See Method 1.
PROCEDURE
Extraction
7.—7.1 Weigh to the nearest 0.001 g, 2.5 g of the prepared sample and place it in a dry Kjeldahl flask. Add 15 ml water and stir so as to suspend the substance. Add 20 ml nitric acid (4.2) and carefully add 30 ml sulphuric acid (4.1). When the initial violent reaction has ceased, slowly bring the contents of the flask to boiling and boil for 30 minutes. Allow to cool and then carefully add with mixing about 150 ml water and boil for 15 minutes.
Cool completely and transfer the liquid quantitatively to a 500 ml graduated flask. Make up to volume, mix and filter through a dry fluted filter, discarding the first portion of the filtrate.
Determination
7.2 Determine the phosphorus according to Method 10 on an aliquot part of the clear filtrate.
9b.EXTRACTION OF PHOSPHORUS BY 2% FORMIC ACID
SCOPE
1. This method is for the determination of phosphorus soluble in 2% formic acid (20 g per litre).
FIELD OF APPLICATION
2. Soft natural phosphate exclusively.
PRINCIPLE
3. To differentiate between hard natural phosphates and soft natural phosphates, phosphorus soluble in formic acid is extracted in specific conditions.
REAGENTS
4.—4.1 Formic acid, 2% (20 g per litre): dilute 82 ml formic acid (concentration 98-100% ; d = 1.22 g/ml) to 5 litres with distilled water.
APPARATUS
5.—5.1 500 ml graduated flask (for example Stohmann).
5.2 Rotary shaker, 35-40 turns per minute.
PREPARATION OF THE SAMPLE
6. See Method 1.
PROCEDURE
Extraction
7.—7.1 Weigh to the nearest 0.001 g, 5 g of the prepared sample and place it in a dry 500 ml graduated Stohmann flask (5.1) with a wide neck. While continuously rotating the flask by hand, add the formic acid (4.1) (at 20±1°C) until it is approximately 1 cm below the graduation mark and make up to the volume. Close the flask with rubber stopper and shake for 30 minutes at 20±2°C on the rotary shaker (5.2). Filter the solution through a dry fluted filter, into a dry receiver, discarding the first portion of the filtrate.
Determination
7.2 Determine the phosphorus according to Method 10 in an aliquot part of the clear filtrate.
9c.EXTRACTION OF PHOSPHORUS BY 2% CITRIC ACID
SCOPE
1. This method is for the determination of phosphorus soluble in 2% citric acid (20 g per litre).
FIELD OF APPLICATION
2. Only applicable to basic slag fertilisers in Group 2(a) of Section A and Groups 1, 2 and 4 of Section B of the Table in Schedule 1 of the Fertilisers Regulations 1990(17).
PRINCIPLE
3. Extraction of phosphorus from the fertiliser with a 2% citric acid solution (20 g per litre) in given conditions.
REAGENT
4.—4.1 2% citric acid solution (20 g per litre), prepared from citrci acid and monohydrate.
Note: Verify the concentration of this citric acid solution by titrating 10 ml of the latter with a sodium hydroxide standard solution 0.1 N, using phenolpthalein as an indicator. If the solution is correct, the titre should be 28.55 ml.
APPARATUS
5.—5.1 Rotary shaker: 35-40 turns per minute.
PREPARATION OF THE SAMPLE
6. The analysis is carried out on the product as received after carefully mixing the original sample to ensure it is homogenous. See Method 1.
PROCEDURE
Extraction
7.—7.1 Weigh to the nearest 0.001 g, 5 g of the prepared sample and plcae it in a dry flask with a sufficiently wide neck, with a capacity of 600 ml, allowing the liquid to be shaken thoroughly. Add 500 ml±1 ml of the citric acid solution (4.1) at 20±1°C. When adding the first mls of the reagent shake vigorously by hand to stop the formation of lumps and to prevent the substance sticking to the sides. Close the flask with a rubber stopper and shake it on the rotary shaker (5.1) for exactly 30 minutes at a temperature of 20±2°C.
Filter immedsiately through a dry fluted filter, into a dry glass receiver and discard the first 20 ml of the filtrate. Continue the filtering until a sufficient quantity of filtrate is obtained to carry out the phosphorus determination.
Determination
7.2 Determine the phosphorus according to Method 10 on an aliquot part of the clear filtrate.
9d.EXTRACTION OF PHOSPHORUS BY NEUTRAL AMMONIUM CITRATE
SCOPE
1. This method is for the determination of phosphorus soluble in neutral ammonium citrate.
FIELD OF APPLICATION
2. All fertilisers in Group 2(a) of Section A and Groups 1, 2 and 4 of Section B and Group 2 of Section C of the Table in Schedule 1 of the Fertilisers Regulations 1990(18) in respect of which solubility in neutral ammonium citrate is laid down.
PRINCIPLE
3. Extraction of phosphorus at a temperature of 65°C using a neutral ammonium citrate solution (pH=7.0) under specific conditions.
REAGENTS
4.—4.1 Neutral ammonium citrate solution (pH=7.0).
This solution must contain per litre 185 g of citric acid monhydrate and must have a specific gravity of 1.09 at 20°C and a pH of 7.0. The reagent is prepared as follows:
dissolve 370 g citric acid monohydrate in about 1.5 litres of water and make an approximately neutral solution by adding 345 ml of ammonia solution (28-29% of NH3). If the NH3 concentration is lower than 28% add a correspondingly larger quantity of ammonia solution and dilute the citric acid in correspondingly smaller quantities of water.
Cool and make exactly neutral by keeping the electrodes of the pH meter (5.1) immersed in the solution, add the ammonia solution (28-29% of NH3) drop by drop, stirring continuously (with a mechanical stirrer) until a pH of exactly 7.0 at 20°C is obtained.
At this point make up the volume to 2 litres and test the pH again. Keep the reagent in a closed container and check the pH at regular intervals.
APPARATUS
5.—5.1 pH meter.
5.2 Water bath which can be set thermostaically at 65°C, equipped with a mechanically operated shaking tray (see Figure 8 in the Appendix).
PREPARATION OF THE SAMPLE
6. See Method 1.
PROCEDURE
Extraction
7.—7.1 Transfer 1(19) or 3(20) grams, as appropriate, of the fertiliser to be analysed into a 200 or 250 ml Erlenmeyer flask containing 100 ml of ammonium citrate solution previously heated to 65°C.
Stopper the Erlenmeyer flask and shake in order to suspend the fertiliser without forming lumps. Remove the stopper for an instant in order to balance the pressure and close the Erlenmeyer flask again. Place the flask in the water-bath (5.2) set to maintain the contents of the flask at exactly 65°C. Shake mechanically for one hour so as to ensure complete suspension of the sample(21). The level of suspension in the flask must stay constantly below that of the water in the bath. After exactly one hour remove the Erlenmeyer flask from the water-bath. Cool immediately under running water to ambient temperature and quantitatively transfer the contents from the Erlenmeyer flask into a graduated 500 ml flask with a jet of water. Make up the volume with water. Mix thoroughly and filter through a dry fluted filter (medium speed) into a dry container, discarding the first part of the filtrate (about 50 ml).
About 100 ml of clear filtrate should be collected.
Determination
7.2 Determine the phosphorus according to Method 10 in an aliquot part of the clear filtrate.
9e.EXTRACTION OF PHOSPHORUS BY ALKALINE AMMONIUM CITRATE (PETERMANN'S METHOD) AT 65°C.
SCOPE
1. This method is for the determination of phosphorus soluble in alkaline ammonium citrate.
FIELD OF APPLICATION
2. Exclusively to precipitated dihydrated dicalcium phosphate (CaHPO4.2H2O).
PRINCIPLE
3. Extraction of phosphorus at a temperature of 65°C with an alkaline solution of ammonium citrate (Petermann) under specified conditions.
REAGENTS
Petermann’s solution
Preparation from diammonium citrate:
Dissolve 941 g diammonium citrate in about 3,500 ml water in a 5 litre graduated flask. Stand the flask in a bath of running water, mix and cool. Add, in small amounts, 430 ml of ammonia solution (d = 0.880 g/ml), from a freshly opened bottle (or an equivalent amount of diluted ammonia, for example if d = 0.906 g/ml then 502 ml are required). Adjust the temperature to 20°C, make up to volume with water and mix.
Preparation from citric acid and ammonia:
Dissolve 865 g citric acid monohydrate in about 2,500 ml distilled water in a container of about 5 litres capacity. Place container in an ice bath and add in small amounts, shaking continually, 966 ml of ammonia solution (d = 0.880 g/ml), from a freshly opened bottle (or an equivalent amount of diluted ammonia, for example if d = 0.906 g/ml, then, 1,114 ml are required). Adjust the temperature to 20°C, transfer to a 5 litre graduated flask, make up to the mark with distilled water and mix.
Check the ammoniacal nitrogen content as follows:
Transfer 25 ml of the solution into a 250 ml graduated flask, make up to volume with distilled water and mix. Determine the ammoniacal nitrogen content on 25 ml of this solution following Method 2. If the solution is correct, 15 ml 0.5 N H2SO4 are consumed—Calculate the concentration of ammoniacal nitrogen in the reagent solution (1 ml 0.5 N H2SO4 = 0.007 g nitrogen).
If the concentration of ammoniacal nitrogen is greater than 42 g/litre, ammonia can be expelled by a stream of inert gas or by moderate heating to bring back the pH to 9.7. Carry out a second determination.
If the concentration of ammoniacal nitrogen is less than 42 g/litre, calculate the volume of ammonia solution required to achieve this level (1 ml ammonia solution, d = 0.880 g/ml contains approximately 0.22 g ammoniacal nitrogen). For each ml of ammonia solution required add 0.173 g of citric acid.
Whenever corrections are made to this reagent solution, it is imperative that the final concentration of both the citric acid and ammoniacal nitrogen are as specified.
APPARATUS
5.—5.1 Water bath which can be maintained at a temperature of 65°±1deg;C.
5.2 500 ml graduated flask (for example Stohmann flask).
PREPARATION OF SAMPLE
6. See Method 1.
PROCEDURE
Extraction
7.—7.1 Weigh to the nearest 0.001 g, 1 g of the prepared sample and transfer to the 500 ml graduated flask (5.2). Add 200 ml alkaline ammonium citrate solution (4.1). Stopper the flask and shake vigorously by hand to avoid the formation of lumps and to prevent any adherence of the substance to the sides.
Place the flask in the water bath at 65°C and shake every 5 minutes during the first half an hour. After each shaking, raise the stopper to equilibrate the pressure. The level of water in the water bath should be above the level of solution in the flask. Allow the flask to remain in the water bath a further hour at 65°C and shake every ten minutes. Remove the flask, cool to a temperature of about 20°C, make up to volume of 500 ml with water. Mix and filter through a dry fluted filter paper, rejecting the first portion of filtrate.
Determination
7.2 Determine the phosphate according to Method 10 on an aliquot part of the clear filtrate.
9f.EXTRACTION OF PHOSPHORUS BY ALKALINE AMMONIUM CITRATE (PETERMANN'S METHOD) AT AMBIENT TEMPERATURE
SCOPE
1. This method is for the determination of phosphorus soluble in cold alkaline ammonium citrate.
FIELD OF APPLICATION
2. Disintegrated phosphates exclusively.
PRINCIPLE
3. Extraction of phosphorus at a temperature about 20°C with an alkaline solution of ammonium citrate (Petermann’s solution) in specific conditions.
REAGENT
4. See Method 9e.
APPARATUS
5.—5.1 250 ml graduated flask (for example Stohmann).
5.2 Rotary shaker, 35-40 turns per minute.
PREPARATION OF THE SAMPLE
6. See Method 1.
PROCEDURE
Extraction
7.—7.1 Weigh to the nearest 0.001 g, 2.5 g of the prepared sample and put it in a 250 ml graduated flask (5.1). Add a little of Petermann’s solution (4) at 20°C, shake very hard in order to stop the formation of lumps and to prevent any of the substance adhering to the side of the flask. Make up to the mark with Petermann’s solution and close the flask with a rubber stopper.
Shake for two hours on the rotary shaker (5.2). Filter immediately through a dry fluted filter, into a dry container, discarding the first portion of the filtrate.
Determination
7.2 Determine the phosphorus according to Method 10 on an aliquot part of the clear filtrate.
9g.EXTRACTION OF PHOSPHORUS BY JOULIE'S ALKALINE AMMONIUM CITRATE
SCOPE
1. This method is for the determination of phosphorus soluble in Joulie’s alkaline ammonium citrate.
FIELD OF APPLICATION
2. All the straight and compound phosphate fertilisers, in which the phosphate occurs in an aluminocalcic form.
PRINCIPLE
3. Extraction by shaking vigorously with an alkaline solution of ammonium citrate of defined specification (and where appropriate in the presence of oxine), at about 20°C.
REAGENTS
Joulie’s alkaline solution of ammonium citrate:
4.—4.1 this solution contains 400 g of citric acid monohydrate and 153 g of NH3 per litre. Its free ammonia content is approximately 55 g per litre. It can be prepared by one of the methods described below:
4.1.1In a litre graduated flask, dissolve 400 g of citric acid monohydrate in approximately 600 ml ammonia solution (d = 0.925 g/ml), containing 200 g NH3 per litre; this may be prepared by diluting 760 ml ammonia solution (d = 0.880 g/ml) from a freshly opened bottle with water to 1 litre. The citric acid is added successively in quantities of 50 to 80 g maintaining the temperature below 50°C. Make up the volume to 1 litre with ammonia.
4.1.2In a 1 litre graduated flask, dissolve 400 g of diammonium citrate in 300 ml of water. Add 440 ml of ammonia solution (d = 0.925 g/ml) (see 4.1.1 above). Make up the volume to 1 litre with water.
Verification of the total ammonia content:
take a 10 ml sample of the citrate solution and place it in a 250 ml flask. Make up to the volume with distilled water. Determine the ammoniacal nitrogen content on 25 ml of this solution according to Method 2. In these conditions the reagent is considered to be correct when the volume of 0.5 N sulphuric acid consumed is between 17.7 and 18.0 ml (1 ml 0.5 N H2SO4-0.008516 g NH3). If this is not so add 4.25 ml of ammonia (d = 0.925 g/ml) per 0.1 ml below the 18 ml indicated above.
4.2 8-Hydroxyquinoline (oxine), powdered.
APPARATUS
5.—5.1 Rotary shaker, 35-40 turns per minute.
PREPARATION OF THE SAMPLE
6. See Method 1.
PROCEDURE
Extraction
7.—7.1 Weigh to the nearest 0.0005 g, 1 g of the prepared sample and place in a small mortar (glass or porcelain). Add about ten drops of ammonium citrate solution (4.1) to moisten it and break it up very carefully with a pestle. Add 20 ml ammonium citrate solution (4.1), mix to a paste and leave it to settle for about 1 minute.
Decant the liquid in to a 500 ml graduated flask straining off particles which might have escaped the preceding moist disintegration. Add 20 ml ammonium citrate solution (4.1) to the residue, grind as above and decant the liquid into the graduated flask. Repeat the process four times, so that by the end of the fifth time all the product can be poured into the flask. The total quantity of ammonium citrate solution used for these processes must be approximately 100 ml.
Rinse the pestle and mortar above the graduated flask with 40 ml of distilled water.
Stopper the flask and shake for three hours on the rotary shaker (5.1).
Leave the flask standing for fifteen to sixteen hours and then shake it again under the same conditions for three hours. The temperature during the whole process should be kept at 20°±2°C.
Make up to volume with distilled water and mix. Filter through a dry filter, discard the first portion of the filtrate and collect the clear filtrate in a dry flask.
Determination
7.2 Determine the phosphorus according to Method 10 on an aliquot part of the clear filtrate.
APPENDIX
8. The use of oxine makes it possible to apply this method to fertilisers containing magnesium. This is recommended when the ratio of magnesium and phosphoric anhydride contents is higher than 0.03 (Mg/P2O5>0.03). If this is the case, add 3 g of oxine to the moistened sample for analysis. The use of oxine in the absence of magnesium is not, moreover, likely to interfere subsequently with the determination. In the known absence of magnesium, oxine may be omitted.
9h.EXTRACTION OF PHOSPHORUS BY WATER
SCOPE
1. This method is for the determination of water-soluble phosphorus.
FIELD OF APPLICATION
2. All fertilisers, including compound fertilisers, where water-soluble phosphorus is to be determined.
PRINCIPLE
3. Extraction in water by shaking under specific conditions.
APPARATUS
4.—4.1 500 ml graduated flask (for example Stohmann).
4.2 Rotary shaker, 35-40 turns per minute.
PREPARATION OF THE SAMPLE
5. See Method 1.
PROCEDURE
Extraction
6.—6.1 Weigh to the nearest 0.001 g, 5 g of the prepared sample and place it in a 500 ml graduated flask (4.1). Add to the flask 450 ml of water, the temperature of which must be between 20° and 25°C. Shake on the rotary shaker (4.2) for 30 minutes. Then make up to the mark with water, mix thoroughly by shaking the filter through a dry fluted filter, into a dry container.
Determination
6.2 Determine the phosphorus according to Method 10, on an aliquot part of the clear filtrate.
10.DETERMINATION OF EXTRACTED PHOSPHORUS (Gravimetric method using quinoline phosphomolybdate)
SCOPE
1. This method is for the determination of phosphorus in the extracts from fertilisers.
FIELD OF APPLICATION
2. The method is applicable to all extracts of fertilisers(22), for the determination of the different forms of phosphorus.
PRINCIPLE
3. After hydrolysis, phosphorus is precipitated in an acid solution in the form of quinoline phosphomolybdate. The precipitate is collected, washed, dried at 250°C and weighed.
In the above conditions, compounds likely to be found in the solution (mineral and organic acids, ammonium ions, soluble silicates, etc….) will not interfere provided that a reagent based on sodium molybdate or ammonium molybdate is used in the precipitation.
REAGENTS
4.—4.1 Concentrated nitric acid (d = 1.40 g/ml).
4.2 Molybdate reagent:
4.2.1Preparation of the reagent based on sodium molybdate:
Solution A: dissolve 70 g sodium molybdate dihydrate in 100 ml water.
Solution B: dissolve 60 g citric acid monohydrate in 100 ml water and add 85 ml concentrated nitric acid (4.1).
Solution C: stir solution A into solution B to obtain solution C.
Solution D: to 50 ml water add 35 ml concentrated nitric acid (4.1), add 5 ml freshly distilled quinoline. Add this solution to solution C, mix thoroughly and leave standing overnight in the dark. Make up to 500 ml with water, mix again and filter through a sintered glass funnel (5.3).
4.2.2Preparation of the reagent based on ammonium molybdate:
Solution A: dissolve 100 g ammonium molybdate in 300 ml water, heating gently and stirring from time to time.
Solution B: dissolve 120 g citric acid monohydrate in 200 ml water and add 170 ml of concentrated nitric acid (4.1).
Solution C: add 10 ml freshly distilled quinoline to 70 ml of concentrated nitric acid (4.1).
Solution D: slowly pour, stirring well, solution A into solution B. After thoroughly mixing, add solution C to this mixture and make up to 1 litre with water. Leave standing for two days in a dark place and filter through a sintered glass funnel (5.3).
The reagents 4.2.1 and 4.2.2 can be used in the same way; both must be kept in the dark in stoppered polyethylene bottles.
APPARATUS
5.—5.1 Filter crucible with porosity of 5 to 20 microns.
5.2 Drying oven regulated at 250°C±10°C.
5.3 Sintered glass funnel with porosity of 5 to 20 microns.
PROCEDURE
Treatment of the solution
6.—6.1 With a pipette take an aliquot part of fertiliser extract (see the Table) containing about 0.01 g of P2O5 and put it in a 500 ml Erlenmeyer flask. Add 15 ml concentrated nitric acid(23) (4.1) and dilute with water to about 100 ml.
Hydrolysis
6.2 Bring the contents of the Erlenmeyer flask to the boil slowly and keep at this temperature until hydrolysis is completed (this usually takes 1 hour). Care must be taken to avoid losses by splashing and excessive evaporation which would reduce the initial volume by more than half, by fitting a reflux condenser. After hydrolysis make up to the initial volume with distilled water.
Weighing the crucible
6.3 Dry the filter crucible (5.1) for at least 15 minutes in the drying oven (5.2). Cool the crucible in a desiccator and weigh.
Precipitation
6.4 Heat the acid solution in the Erlenmeyer flask until it begins to boil and then precipitate the quinoline phosphomolybdate by adding 40 ml of the precipitating reagent (4.2.1 or 4.2.2)(24) drop by drop, stirring continuously. Place the Erlenmeyer flask in a steam bath, leave it there for 15 minutes, shaking it from time to time. The solution can be filtered immediately or after it has cooled down.
Filtering and Washing
6.5 Filter the solution under vacuum by decantation. Wash the precipitate in the Erlenmeyer flask with 30 ml water. Decant and filter the solution. Repeat this process five times. Quantitatively transfer the rest of the precipitate into the crucible washing it with water. Wash four times with 20 ml water, allowing the liquid to drain from the crucible before each addition.
Drying and weighing
6.6 Wipe the outside of the crucible with a filter paper. Place the crucible in the drying oven (5.5) and keep it there until its weight remains constant at a temperature of 250°C (usually 15 minutes); leave it to cool in a desiccator to ambient temperature and weigh rapidly.
Blank test
6.7 For each series of determination, make a blank test under the same conditions (omitting only the sample) and allow for this in the calculation of the final result.
Control test
6.8 Carry out the determination using an aliquot part of a potassium dihydrogen phosphate solution containing 0.01 g of P2O5.
TABLE FOR METHOD 10
| %P205 in the fertiliser | %P in the fertiliser | Sample for analysis g | Dilution to ml | Sampl ml | Dilution to ml | Sample to be precipitated ml | Quinoline phosphomolybdate conversion factor (F) in percentage P205 | Quinoline phosphomolybdate conversion factor (F) in percentage P |
|---|---|---|---|---|---|---|---|---|
| 1 | 500 | — | — | 100 | 16.037 | 6.992 | ||
| 1-5 | 0.44-2.2 | 2.5 | 500 | — | — | 50 | 12.830 | 5.594 |
| 5 | 500 | — | — | 25 | 12.830 | 5.594 | ||
| 1 | 500 | — | — | 50 | 32.074 | 13.984 | ||
| 5-10 | 2.2-4.4 | 2.5 | 500 | — | — | 25 | 25.660 | 11.188 |
| 3 | 500 | — | — | 25 | 21.383 | 9.323 | ||
| 5 | 500 | — | — | 10 | 32.074 | 13.984 | ||
| 1 | 500 | — | — | 25 | 64.148 | 27.968 | ||
| 10-25 | 4.4-11.0 | 2.5 | 500 | — | — | 10 | 64.148 | 27.968 |
| 3 | 500 | — | — | 10 | 53.457 | 23.307 | ||
| 5 | 500 | — | — | 50 | 64.148 | 27.968 | ||
| 1 | 500 | — | — | 10 | 160.370 | 69.921 | ||
| +25 | +11 | 2.5 | 500 | 50 | 500 | 50 | 128.296 | 55.937 |
| 3 | 500 | 50 | 500 | 50 | 106.913 | 46.614 | ||
| 5 | 500 | 50 | 500 | 25 | 128.296 | 55.937 |
EXPRESSION OF RESULTS
7. If the samples for analysis and diltutions shoen in the Table are used the following formulae apply:
% P in the fertiliser = (A − a) × F’
% P2O5 in the fertiliser = (A − a) × F
where:
A = weight in g of the quinoline phosphomolybdate
a = weight in g of the quinoline phosphomolybdate obtained in the blank test.
F and F’ = factors given in the last two columns of the Table.
With samples for analysis and dilutions which differ from those of the Table the following formulae apply:
where:
f = conversion factor, quinoline phosphomolybdate into P2O5 = 0.032074
f’ = conversion factor, quinoline phosphomolybdate into P = 0.013998
D = dilution factor
M = weighing of the sample analysed.

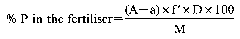
11.DETERMINATION OF WATER-SOLUBLE POTASSIUM
SCOPE
1. This method is for the determination of water-soluble potassium.
FIELD OF APPLICATION
2. All the potassium fertilisers listed in Group 3(a) of Section A and Groups 1, 3 and 4 of Section B and Group 2 of Section C of the Table in Schedule 1 of the Fertilisers Regulations 1990(25).
PRINCIPLE
3. The potassium is extracted with water and after the removal of interfering substances, the potassium is precipitated in a slightly alkaline medium in the form of potassium tetraphenylborate (KTPB).
REAGENTS
4.—4.1 Formaldehyde, 25-30% solution, filter if necessary before use.
4.2 Potassium chloride.
4.3 Sodium hydroxide, 10 N solution. Care should be taken to ensure that the sodium hydroxide is free from potassium.
4.4 Indicator solution: dissolve 0.5 g phenolphtalein in 100 ml 90% ethanol.
4.5 EDTA solution: 4 g of the dihydrated disodium salt of ethylenediaminetetra-acetic acid (EDTA) per 100 ml. Store this reagent in a plastic container.
4.6 STPB solution: dissolve 32.5 g sodium tetraphenylborate in 480 ml of water, add 2 ml sodium hydroxide solution (4.3) and 20 ml of a magnesium chloride solution (100 g of MgCL2.6H2O per litre). Stir for fifteen minutes and filter through a fine, ashless filter. Store this reagent in a plastic container.
4.7 Liquid for washing: dilute 20 ml of the STPB solution (4.6) to 1 litre with water.
4.8 Bromine water: saturated bromine solution in water.
APPARATUS
5.—5.1 Filter crucibles with a porosity of 5 to 20 microns.
5.2 Oven regulated at 120±10°C.
PREPARATION OF THE SAMPLE
6. See Method 1.
In the case of potassium salts the sample must be ground fine enough in order that a representative sample is obtained for analysis. For these products, Method 1, paragraph 6(a) must be used.
PROCEDURE
Extraction
7.—7.1 Weigh to the nearest 0.001g, 10 g of the prepared sample (5 g for potassium salts containing more than 50% of potassium oxide or 20 g for fetilisers containing less than 5% of potassium oxide) and place in a 600 ml beaker with approximately 400 ml of water. Bring to the boil and allow it to boil for 30 minutes. Cool, transfer quantitatively into a 1 litre graduated flask, make up the volume, mix and filter into a dry receiver. Discard the first 50 ml of the filtrate.
Note: If the filtrate is dark in colour, transfer by pipette, an aliquot part containing at the most 100 mg of K2O and place in a 100 ml graduated flask, add bromine water and bring to the boil to eliminate any surplus bromine. After cooling make up the volume, filter and quantitatively determine the potassium in an aliquot part of the filtrate.
Determination
7.2 Transfer by pipette an aliquot part of the filtrate containing 25-50 mg of potassium (see Table on page 53) into a 250 ml beaker; make up to 50 ml with water.
To remove interferences, add 10 ml of the EDTA solution (4.5), several drops of the phenolphthalein solution (4.4) and stir in, drop by drop, sdoium hydroxide solution (4.3) until it turns red, then finally add a few more drops of sodium hydroxide to ensure an excess (usually 1 ml of sodium hydroxide is sufficient to neutralise the sample and ensure an excess).
To eliminate most of the ammonia boil gently for 15 minutes. Add water to make the volume up to 60 ml.
Bring the solution to the boil, remove the beaker from the heat and add 10 ml formaldehyde (4.1). Add several drops of phenolphthalein solution (4.4) and if necessary, more sodium hydroxide solution until a distinct red colour appears. Cover the beaker with a watch glass and place it on a steam bath for fifteen minutes.
Weighing the crucible
7.3 Dry the filter crucible (5.1) to constant weight in the oven at 120°C (5.2) (about 15 minutes).
Allow the crucible to cool in a desiccator and then weigh it.
Precipitation
7.4 Remove the beaker from the steam bath, stir in drop by drop 10 ml of the STPB solution (4.6). This addition should take about 2 minutes; allow to stand for at least 10 minutes before filtering.
Filtering and washing
7.5 Filter under vacuum into the weighed crucible, rinse the beaker with the liquid for washing (4.7), wash the precipitate three times with the liquid for washing (60 ml in all of the liquid for washing) and twice with 5 to 10 ml of water.
Drying and weighing
7.6 Wipe the outside of the crucible with a filter paper and place in the oven (5.2) for one and a half hours at temperature of 120°C. Allow the crucible to cool in a desiccator to ambient temperature and weigh rapidly.
Blank test
7.7 Make a blank test under the same conditions (omitting only the sample) and allow for this in the calculation of the final result.
Control test
7.8 Carry out the determination of an aliquot part of an aqueous solution of potassium chloride, containing at the most 40 mg of K2O.
EXPRESSION OF RESULTS
Method of calculation and formulae
8.—8.1 If the quantities and the dilutions shown in the Table are used, the following formulae apply:
% K2O in the fertiliser = (a − a) × F
or
% K in the fertiliser = (A − a) × F’
where:
A = weight in grams of the precipitate from the sample
a = weight in grams of the precipitate from the blank
F and F’ = factors—see Table.
% of K2O in the fertiliser % of K in the fertiliser Sample for analysis (g) Aliquot part to be taken as a sample for precipitation (ml) Conversion factor F % K2O g KTPB Conversion factor F % K g KTPB 1 — 5 0.8 — 4.2 20 50 13.140 10.906 5 — 10 4.2 — 8.3 10 50 26.280 21.812 10 — 20 8.3 — 16.6 10 25 52.560 43.624 20 — 50 16.6 — 41.5 10 10 131.400 109.060 more than 50 more than 41.5 5 10 262.800 218.120 With samples and dilutions which differ from those shown in the Table use the following formulae:
or
where:
f = conversion factor, KTPB into K2O = 0.1314
f’ = conversion factor, KTPB into K = 0.109
D = dilution factor
M = weight in grams of sample for analysis

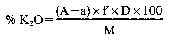
12a.DETERMINATION OF WATER-SOLUBLE MAGNESIUM—ATOMIC ABSORPTION SPECTROPHOTOMETRIC METHOD
SCOPE
1. This method is for the determination of water-soluble magnesium.
FIELD OF APPLICATION
2. Exclusively to fertilisers in Groups 1(a) and 3(a) of Section A of the Table in Schedule 1 of the Fertilisers Regulations 1990(26), in respect of which the declaration of water-soluble magnesium is required.
PRINCIPLE
3. Solution of magnesium by boiling the test sample in water, and determination by atomic absorption spectrophotometry.
REAGENTS
4.—4.1 Hydrochloric acid, N solution (approximately).
4.2 Hydrochloric acid, 0.5 N solution.
4.3 Magnesium standard solution: dissolve 1.013 g magnesium sulphate (MgSO4.7H2O) in 0.5 N hydrochloric acid solution (4.2) and dilute to 100 ml with this acid.
1 ml of this solution contains 1 mg of magnesium (Mg).
or
weigh out 1.658 g of magnesium oxide, previously calcined at 600° for 2 hours, place in a beaker with 100 ml of water and 120 ml of approximately N hydrochloric acid (4.1). After dissolution, transfer quantitively into a one litre graduated flask, make up the volume with water and mix.
1 ml of this solution contains 1 mg of magnesium (Mg).
4.4 Strontium chloride solution: dissolve 15 g strontium chloride (SrCl2.6H2O) in 0.5 N hydrochloric acid solution (4.2) and dilute to 100 ml with the same solvent.
APPARATUS
5.—5.1 Atomic absorption spectrophotometer with a magnesium lamp (285.2 nm).
PREPARATION OF SAMPLE
6. See Method 1.
PROCEDURE
Extraction
7.—7.1 Weigh to the nearest 0.001 g, 5 g of the prepared sample and place in a 500 ml graduated flask. Add about 300 ml water and boil for half an hour. Allow to cool, dilute to the mark with water, mix and filter.
Preparation of the sample solution
7.2 7.2.1 If the fertiliser has a declared magnesium oxide (MgO) content greater than 10%, transfer by pipette 25 ml of the filtrate (7.1) into a 100 ml graduated flask, make up to the mark with water and mix.
7.2.2Transfer by pipette 10 ml of the filtrate (7.1) or the diluted filtrate (7.2.1) into a 200 ml graduated flask and make up to the mark with 0.5 N hydrochloric acid solution (4.2).
7.2.3Dilute solution (7.2.2) with 0.5 N hydrochloric acid solution (4.2) to a concentration within the working range of the spectrophotometer.
The final solution must contain 10% V/V of the strontium chloride solution (4.4).
Blank solution
7.3 Prepare a blank solution from which only the sample has been omitted.
Standard solutions for calibration
7.4 By diluting the standard solution (4.3) with 0.5 N hydrochloric acid solution (4.2), prepare at least 5 standard solutions of increasing concentration corresponding to the optimal measuring range of the spectrophotometer. The final solutions must contain 10% V/V of the strontium chloride solution (4.4).
Measurement
7.5 Set up the spectrophotometer (5.1), at a wavelength of 28.25 nm using an oxidising air-acetylene flame. Spray successively, in triplicate, the standard solutions (7.4), the sample solution (7.2) and the blank solution (7.3), washing the instrument through with distilled water between each spraying. Plot the calibration curve using the mean absorbances as the ordinates and the corresponding concentrations of magnesium in Ö as the abscissae. Determine the concentration of magnesium in the sample and blank by reference to the claibration curve.
EXPRESSION OF RESULTS
8. Calculate the quantity of magnesium (Mg) or magnesium oxide (MgO) (conversion factor Mg to MgO = 1.66) in the sample taking into consideration the blank. Express the result as a percentage of the sample.
12b.DETERMINATION OF WATER-SOLUBLE MAGNESIUM—EDTA METHOD
SCOPE
1. This method is for the determination of water-soluble magnesium.
FIELD OF APPLICATION
2. Exclusively to straight fertilisers in Groups 1(a) and 3(a) of Section A of the Table in Schedule 1 of the Fertilisers Regulations 1990(27) in respect of which the indication of water-soluble magnesium, expressed as magnesium oxide, is required.
PRINCIPLE
3. Solution of magnesium by boiling a test sample in water. Titration with EDTA of calcium and magnesium in the presence of eriochrome black-T, followed by titration with EDTA of calcium in the presence of calcein or of calcon carbonic acid. Determination of magnesium by difference.
REAGENTS
4.—4.1 Magnesium solution, 0.05 M: weigh out 2.016 g magnesium oxide, previously calcined at 600°C for 2 hours, place in a beaker with 100 ml water and stir in 120 ml of approximately N hydrochloric acid. After dissolution, transfer quantitatively into a 1 litre graduated flask, make up the volume with water and mix. Check the strength of the solution gravimetrically by precipitation as magnesium ammonium phosphate.
1 ml of the solution should contain 1.216 mg of Mg (= 2.016 mg of MgO).
4.2 EDTA solution, 0.05 M: dissolve 18.61 g of the dihydrated disodium salt of ethylenediaminetetra-acetic acid in 600-800 ml water contained in a 1 litre beaker. Transfer the solution quantitatively to a 1 litre graduated flask, make up to volume with water and mix. Check this solution (4.1) by taking a sample of 20 ml of the latter and titrating as described under 7.3.1.
1 ml of the EDTA solution should correspond to 1.216 mg of Mg or 2.016 mg of MGO and to 2.004 mg of Ca or 2.804 mg of CaO.
4.3 Calcium solution 0.05 M: weigh out 5.004 g of dry calcium carbonate and place in a beaker with 100 ml water. Progressively stir in 120 ml approximately N hydrochloric acid. Bring to the boil in order to drive off the carbon dioxide, cool, transfer quantitatively into a 1 litre graduated flask, make up to volume with water and mix. Check the solution against the EDTA solution (4.2) following analytical procedure 7.3.2. One ml of this solution should contain 2.004 mg of Ca (= 2.804 mg of CaO) and should correspond to 1 ml of the 0.05 molar EDTA solution.
4.4 Calcein indicator: carefully mix in a mortar 1 g calcein with 100 g sodium chloride. Use 10 mg of this mixture. The indicator changes from green to orange. Titration must be carried out until an orange colour is obtained which is free from green tinges.
4.5 Calcon carbonic acid indicator: dissolve 400 mg calcon carbonic acid in 100 ml methanol. Use three drops of this solution. The indicator changes from red to blue. Titration must be carried out until a blue colour is obtained which is free from red tinges.
4.6 Eriochrome black-T indicator: dissolve 300 mg eriochrome black-T in a mixture of 25 ml propan-1-ol and 15 ml triethanolamine. Use three drops of this solution. This indicator turns from red to blue and titration must be carried out until a blue colour is obtained which is free from rred tinges. It changes colour only when magnesium is present. If necessary add 0.1 ml of standard solution 4.1.
4.7 Potassium cyanide solution, 2 g per 100 ml.
4.8 Solution of potassium hydroxide and potassium cyanide: dissolve 280 g potassium hydroxide and 66 g potassium cyanide in water, make up the volume to 1 litre and mix.
4.9 pH 10 buffer solution: dissolve 33 g ammonium chloride in 200 ml water, add 207 ml ammonia solution (d = 0.880 g/ml) from a freshly opened bottle (or an equivalent amount of diluted ammonia, for example if d = 0.91 g/ml, use 250 ml). Make up the volume to 500 ml with water and mix. Check the pH of this solution regularly.
APPARATUS
5.—5.1 Magnetic or mechanical stirrer.
5.2 pH meter.
PREPARATION OF THE SAMPLE
6. See Method 1.
PROCEDURE
Extraction
7.—7.1 Weigh to the nearest 0.001 g, 5 g of the prepared sample and place in a 500 ml graduated flask. Add about 300 ml water and boil for half an hour. Cool, make up the volume, mix and filter.
Control test
7.2 Carry out a determination on aliquot parts of solutions (4.1) and (4.3) such that the Ca/Mg ratio is equal to that expected from the sample. For this purpose take (a) ml of standard solution (4.3) and (b — a) ml standard solution (4.1), where (a) and (b) are the numbers of ml EDTA solution used in the two titrations when analysing the sample. This procedure is correct only if the standard solutions of EDTA, calcium and magnesium are exactly equivalent. If this is not the case, it is necessary to make the appropriate corrections.
Determination
7.3.1 Titration in the presence of eriochrome black-T
7.3.1Place an aliquot part of the solution to be analysed (see the Table) in a 300 ml beaker and dilute with water to about 100 ml. Add 5 ml buffer solution (4.9). The pH measured by the meter (5.2) must be 10.5±0.1. Add 2 ml potassium cyanide solution (4.7) and 3 drops of the eriochrome black-T indicator (4.6). Stir gently and titrate with the EDTA solution (4.2). Let “b” be the number of ml of 0.05 molar EDTA solution.
Note: For titration with eriochrome black-T, the titration must not exceed 25 ml of EDTA, otherwise the volume of the aliquot part must be reduced.
7.3.2 Titration in the presence of calcein or of calcon carbonic acid
7.3.2Place an aliquot part of the solution to be analysed equal to that taken for the above titration in a beaker. Dilute with water to about 100 ml. Add 10 ml potassium hydroxide-potassium cyanide solution (4.8) and the indicator (4.4) or (4.5). Stir gently and titrate with the EDTA solution (4.2). Let “a” be the number of ml of 0.05 molar EDTA solution.
EXPRESSION OF RESULTS
8.


M = weight of the sample, expressed in grams, present in the aliquot part.
TABLE FOR METHOD 12
| Type of fertiliser | Aliquot part to be taken as sample for each titration | Quantity of sample present in one aliquot part |
|---|---|---|
| Nitrate of calcium and of magnesium | 20 ml | 0.200 g |
| Magnesium ammonium sulphate-nitrate | 50 ml | 0.500 g |
| Crude potassium salts | 25 ml | 0.250 g |
| Potassium magnesium chloride | 25 ml | 0.250 g |
| Sulphate of potassium and magnesium | 25 ml | 0.250 g |
13a.DETERMINATION OF TOTAL MAGNESIUM—ATOMIC ABSORPTION SPECTROPHOTOMETRIC METHOD
SCOPE
1. This method is for the determination of total magnesium.
FIELD OF APPLICATION
2. Exclusively to the fertiliser magnesium ammonium nitrate in Group 1(a) of Section A of the Table in Schedule 1 of the Fertilisers Regulations 1990(28) in respect of which the declaration of total magnesium is required.
PRINCIPLE
3. Solution of magnesium by boiling the test sample in dilute acid, and determination by atomic absorption spectrophotometry.
REAGENTS
4.—4.1 Hydrochloric acid solution 50% (V/V): dilute an appropriate volume of hydrochloric acid (d = 1.18 g/ml) with an equal volume of water.
4.2 Hydrochloric acid, N solution (approximately).
4.3 Hydrochloric acid, 0.5 N solution.
4.4 Magnesium solution: dissolve 1.013 g magnesium sulphate (MgSO4.7H2O) in 0.5 N hydrochloric acid solution (4.3) and dilute to 100 ml with this acid.
1 ml of this solution contains 1 mg of magnesium (Mg).
or
weigh out 1.658 g of magnesium oxide, previously calcined at 600°C for 2 hours, place in a beaker with 100 ml of water and 120 ml of approximately N hydrochloric acid (4.2). After dissolution, transfer quantitatively into a one litre graduated flask, make up the volume with water and mix.
1 ml of this solution contains 1 mg of magnesium (Mg).
4.5 Strontium chloride solution: dissolve 75 g strontium chloride (SrCl2.6H2O) in 0.5 N hydrochloric acid solution (4.3) and dilute to 500 ml with this acid.
APPARATUS
5.—5.1 Atomic absorption spectrophotometer with a magnesium lamp (285.2 nm).
PREPARATION OF SAMPLE
6. See Method 1.
PROCEDURE
Extraction
7.—7.1 Weigh to the nearest 0.001 g, 5 g of the prepared sample and place in a 500 ml graduated flask. Add about 200 ml water, 20 ml hydrochloric acid solution (4.1) and boil for half an hour. Allow to cool, dilute to the mark with water, mix and filter.
Preparation of the sample solution
7.2 7.2.1 If the fertiliser has a declared magnesium oxide (MgO) content greater than 10%, transfer by pipette 25 ml of the filtrate (7.1) into a 100 ml graduated flask, make up to the mark with water and mix.
7.2.2Transfer by pipette 10 ml of the filtrate (7.1) or the diluted filtrate (7.2.1), into a 200 ml graduated flask and make up to the mark with 0.5 N hydrochloric acid solution (4.3).
7.2.3Dilute solution (7.2.2) with 0.5 N hydrochloric acid solution (4.3) to a concentration within working range of the spectrophotometer. The final solution must contain 10% V/V strontium chloride solution (4.5).
Blank solution
7.3 Prepare a blank solution from which only the sample has been omitted.
Standard solutions for calibration
7.4 By diluting the standard solution (4.4) with 0.5 hydrochloric acid solution (4.3), prepare at least 5 standard solutions of increasing concentration corresponding to the optimal measuring range of the spectrophotometer. The final solutions must contain 10% V/V of the strontium chloride solution (4.5).
Measurement
7.5 Set up the spectrophotometer (5.1), at a wavelength of 285.2 nm using an oxidising air-acetylene flame. Spray successively, in triplicate, the standard solutions (7.4), the sample solution (7.2) and the blank solution (7.3), washing the instrument through with distilled water between each spraying. Plot the calibration curve using the mean absorbances as the ordinates and the corresponding concentrations of magnesium in Ö as the abscissae. Determine the concetration of magnesium in the sample and blank by reference to the calibration curve.
EXPRESSION OF RESULTS
8. Calculate the quantity of magnesium (Mg) or magnesium oxide (MgO) (conversion factor Mg to MgO = 1.66) in the sample, taking into consideration the blank. Express the result as a percentage of the sample.
13b.DETERMINATION OF TOTAL MAGNESIUM—EDTA METHOD
SCOPE
1. This method is for the determination of total magnesium.
FIELD OF APPLICATION
2. Exclusively to the fertiliser magnesium ammonium nitrate in Group 1(a) of Section A of the Table in Schedule 1 of the Fertilisers Regulations 1990(29) in respect of which the indication of total magnesium is required.
PRINCIPLE
3. Solution of magnesium by boiling a test sample in dilute acid. Titration of calcium and magnesium with EDTA in the presence of eriochrome black-T, followed by titration with EDTA of calcium in the presence of calcein or of calcon carbonic acid. Determination of magnesium by difference.
REAGENTS
4.—4.1 Magnesium solution, 0.05 M: weigh out 2.016 g of magnesium oxide previously calcined at 600°C for 2 hours, place in a beaker with 100 ml of water and stir in 120 ml of approximately 1 N hydrochloric acid. After dissolution, transfer quantitatively into a 1 litre graduated flask, make up the volume with water and mix. Check the strength of the solution gravimetrically by precipitation as ammonium-magnesium phosphate.
1 ml of the solution should contain 1.216 mg of magnesium (Mg) (= 2.016 mg of magnesium oxide (MgO)).
4.2 EDTA solution 0.05 M: dissolve 18.61 g of the dihydrated disodium salt of ethylenediaminetetra-acetic acid in 600-800 ml water contained in a 1 litre beaker. Transfer the solution quantitatively into a 1 litre graduated flask, make up to volume with water and mix. Check this solution with solution (4.1) by taking a sample of 20 ml of the latter and titrating following analytical procedure 7.3.1.
1 ml of the EDTA solution should correspond to 1.216 mg of Mg or 2.016 mg of MgO and to 2.004 mg of Ca or 2.804 mg of CaO.
4.3 Calcium solution 0.05 M: weigh out 5.004 g of dry calcium carbonate and place in a beaker with 100 ml of water. Progressively stir in 120 ml of approximately N hydrochloric acid. Bring to the boil in order to drive off the carbon dioxide, cool, transfer quantitatively into a 1 litre graduated flask, make up to volume with water and mix. Check this solution against the EDTA solution (4.2) following analytical procedure 7.3.2.
One ml of this solution should contain 2.004 mg of Ca (= 2.804 mg of CaO) and should correspond to 1 ml of the 0.05 molar EDTA solution.
4.4 Calcein indicator: carefully mix in a mortar 1 g of calcein with 100 g of sodium chloride. Use 10 mg of this mixture. The indicator changes from green to orange. Titration must be carried out until an orange colour is obtained which is free from green tinges.
4.5 Calcon carbonic acid indicator: dissolve 400 mg of calcon carbonic acid in 100 ml of methanol. Use three drops of this solution. The indicator changes from red to blue. Titration must be carried out until a blue colour is obtained which is free from red tinges.
4.6 Eriochrome black-T indicator: dissolve 300 mg of eriochrome black-T in a mixture of 25 ml of propan-1-ol and 15 ml of triethanolamine. Use three drops of this solution. This indicator turns from red to blue and titration must be carried out until a blue colour is obtained which is free from red tinges. It changes colour only when magnesium is present. If necessary add 0.1 ml of standard solution (4.1).
4.7 Potassium cyanide solution, 2 g per 100 ml.
4.8 Solution of potassium hydroxide and potassium cyanide: dissolve 280 g potassium hydroxide and 66 g potassium cyanide in water, make up the volume to one litre and mix.
4.9 pH 10.5 buffer solution: dissolve 33 g ammonium chloride in 200 ml of water, add 207 ml ammonia solution (d = 0.880 g/ml) from a freshly opened bottle (or an equivalent amount of diluted ammonia, for example if d = 0.91 g/ml, use 250 ml). Make up to volume to 500 ml with water and mix. Check the pH of this solution regularly.
4.10 Hydrochloric acid solution, 50% (V/V): dilute an appropriate volume of hydrochloric acid (d = 1.18 g/ml) with an equal volume of water.
4.11 Sodium hydroxide solution, 5N.
APPARATUS
5.—5.1 Magnetic or mechanical stirrer.
5.2 pH meter.
PREPARATION OF THE SAMPLE
6. See Method 1.
PROCEDURE
Extraction
7.—7.1 Weigh to the nearest 0.001 g, 5 g of the prepared sample and place in a 500 ml graduated flask. Add about 200 ml water and 20 ml hydrochloric acid (4.10) and boil for half an hour. Cool, make up to volume with water, mix and filter.
Control test
7.2 Carry out a determination on aliquot parts of solutions (4.1) and (4.3) such that the Ca/Mg ratio is equal to that expected from the sample. For this purpose take (a) ml of standard solution (4.3) and (b — a) ml standard solution (4.1), where (a) and (b) are the numbers of ml EDTA solution used in the two titrations when analysiing the sample. This procedure is correct only if the standard solutions of EDTA, calcium and magnesium are exactly equivalent. If this is not the case, it is necessary to make the appropriate corrections.
Determination
7.3.1 Titration in the presence of eriochrome black-T
7.3.1Transfer by pipette 50 ml of the solution to be analysed into a 300 ml beaker. Neutralise the excess acid with the 5 N sodium hydroxide solution (4.11) using the pH meter (5.2). Dilute with water to 100 ml. Add 5 ml buffer solution (4.9). The pH measured by the meter must be 10.5±0.1. Add 2 ml potassium cyanide solution (4.7) and three drops eriochrome black-T indicator (4.6). Titrate with the EDTA solution (4.2), stirring gently with the stirrer (5.1). Let “b” be the number of ml of 0.05 molar EDTA solution.
Note: For titration with eriochrome black-T, the titration must not exceed 25 ml of EDTA otherwise the volume of the aliquot part must be reduced.
7.3.2 Titration in the presence of calcein or of calcon carbonic acid
7.3.2Place an aliquot part of the solution to be analysed equal to that taken for the above titration in a 300 ml beaker. Neutralise the excess acid with 5 N sodium hydroxide solution (4.11) using the pH meter (5.2).
Dilute with water to about 100 ml. Add 10 ml potassium hydroxide—potassium cyanide solution (4.8) and the indicator (4.4) or (4.5). Stir gently and titrate with the EDTA solution. Let “a” be the number of ml of 0.05 molar EDTA solution.
EXPRESSION OF RESULTS
8.
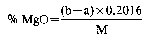

M = weight of the sample, expressed in grams, present in the aliquot part.
14.DETERMINATION OF CHLORIDES IN THE ABSENCE OF ORGANIC MATERIAL
SCOPE
1. This method is for the determination of chloride, in the absence of organic material.
FIELD OF APPLICATION
2. All fertilisers which are free from organic material, except ammonium nitrate fertilisers of a nitrogen content greater than 28% by weight.
PRINCIPLE
3. The chlorides, dissolved in water, are precipitated in an acid medium by an excess of standard solution of silver nitrate. The excess is titrated with a solution of ammonium thiocyanate in the presence of ferric ammonium sulphate (Volhard’s method).
REAGENTS
4.—4.1 Nitrobenzene or diethyl ether.
4.2 Nitric acid, 10 N solution.
4.3 Indictaor solution: dissolve 40 g of ferric ammonium sulphate [Fe2(SO4)3.(NH4)2SO4.24H2O] in water and make up to 1 litre.
4.4 Silver nitrate, 0.1 N solution.
4.5 Ammonium thiocyanate, 0.1 N solution.
Preparation: since this salt is hygroscopic and cannot be dried without risk of decomposition, it is advisable to weigh out approximately 9 g, dissolve in water and make up the volume to one litre. Standardise by titration against 0.1 N silver nitrate solution.
APPARATUS
5.—5.1 Rotary shaker, 35-40 turns per minute.
PREPARATION OF SAMPLE
6. See Method 1.
PROCEDURE
Extraction
7.—7.1 Weigh to the nearest 0.001 g, 5 g of the prepared sample and place in a 500 ml graduated flask and add 450 ml water. Mix for half an hour on the shaker (5.1); make up to 500 ml with distilled water, mix and filter into a beaker.
Determination
7.2 Take an aliquot part of the filtrate containing not more than 0.150 g of chloride. If the sample taken is smaller than 50 ml it is necessary to make up the volume to 50 ml with distilled water. Add 5 ml 10 N nitric acid (4.2), 20 ml indicator solution (4.3), and two drops ammonium thiocyanate standard solution (taken from a burette adjusted to zero). From a burette than add silver nitrate solution (4.4) until there is an excess of 2 to 5 ml. Add 5 ml nitrobenzene or 5 ml diethyl ether (4.1) and shake well to agglomerate the precipitate. Titrate the excess silver nitrate with 0.1 N ammonium thiocyanate (4.5) until a red-brown colour appears which remains after the flask has been shaken slightly.
Note: Nitrobenzene or diethyl ether (especially the former) prevents the silver chloride from reacting with thiocyanate ions, thus a clear colour change is obtained.
Blank test
7.3 Make a blank test under the same conditions (omitting only the sample) and allow for it when calculating the final result.
Control test
7.4 Carry out the determination on an aliquot part of a freshly prepared solution of potassium chloride, containing 0.100 g as chloride.
EXPRESSION OF RESULT
8. Express the result of the analysis as a percentage of chloride contained in the sample as it has been received for analysis.
Calculation: calculate the percentage of chloride (Cl) with the formula:

where:
Vz = number of millilitres of silver nitrate added
Vcz = number of millilitres of silver nitrate used in the blank test
Va = number of millilitres of ammonium thiocyanate used for the titration of the sample
Vca = number of millilitres of ammonium thiocyanate used for the titration of the blank
M = weight in grams of the sample in aliquot volume taken for titration.
15a.DETERMINATION OF FINENESS OF GRINDING—DRY METHOD
SCOPE
1. This method is for the determination of the fineness of grinding by the dry method.
FIELD OF APPLICATION
2. All fertilisers in Schedule 1 of the Fertilisers Regulations 1990(30) for which requirements are given of fineness of grinding using 0.630 mm and 0.160 mm sieves.
PRINCIPLE
3. By mechanical sieve shaking, the quantities of product with a granule size greater than 0.63 mm and those with a granule size between 0.16 mm and 0.63 mm are determined and the percentage of fineness of grinding are calculated.
APPARATUS
4.—4.1 Mechanical sieve shaker.
4.2 Sieves with apertures of 0.160 mm and 0.630 mm respectively of standard ranges (diameter 20 cm, height 5 cm).
PROCEDURE
5. Weigh to the nearest 0.05 g, 50 g of the sample. Assemble the two sieves and the collecting container on the shaker (4.1), the sieve with the larger apertures being placed on top. Place the sample for analysis on the top. Sieve for ten minutes and remove the part collected on the bottom. Sieve again for one minute and check that the amount collected on the bottom during this time is not more than 250 mg. Repeat the process (for one minute each time) until the amount collected is less than 250 mg. Weigh the residual material on both sieves seperately.
EXPRESSION OF RESULTS
6. Percentage of material passing sieve of 0.630 mm apertures = (50 — M1) × 2
Percentage of material passing sieve of 0.160 mm apertures = [50 − (M1 + M2)] × 2
M1 = weight in g of residue on the sieve with 0.630 mm apertures
M2 = weight in g of residue on the sieve with 0.160 mm apertures
The results are to be rounded up to the nearest unit.
15b.DETERMINATION OF THE FINENESS OF GRINDING OF SOFT NATURAL PHOSPHATES
SCOPE
1. This method is for determining the fineness of grinding of soft natural phosphates.
FIELD OF APPLICATION
2. Soft natural phosphates.
PRINCIPLE
3. For samples of fine particle size, agglomeration may occur thus making dry sieving difficult. For this reason, wet sieving is normally used.
REAGENTS
4. Sodium hexametaphosphate solution, 1 g per 100 ml.
APPARATUS
5.—5.1 Sieves with apertures of 0.063 mm and 0.125 mm respectively of standard ranges (diameter 20 cm, height 5 cm) and collecting containers.
5.2 Glass funnel of 20 cm diameter mounted on a stand.
5.3 Laboratory oven.
PROCEDURE
6. Wash both sides of the sieves with water and place the sieve with 0.125 mm apertures above the 0.063 mm sieve.
Weigh to the nearest 0.05 g, 50 g of the prepared sample and place on the top sieve. Sieve under a small jet of cold water (tap water can be used) until the water is practically clear when it passes through. Care should be taken to ensure that the flow of water is such that the lower sieve never fills with water. When the residue on the top sieve seems to remain more or less constant, remove this sieve, and place in the meanwhile on a collecting container.
Continue the wet sieving through the lower sieve for a few minutes, until the water passing through is nearly clear. Replace the 0.125 mm sieve over the 0.063 mm sieve. Transfer any deposit from the collecting container to the top sieve and begin sieving again under a small jet of water until this water becomes almost clear once more.
Quantitatively transfer each of the residues into a seperate 250 ml beaker by means of the funnel. Suspend each residue by filling the beakers with water. Allow to stand for about 1 minute and then decant as much water as possible. Place the beakers in the oven (5.3) at 150°C for two hours. Allow them to cool, detach the residues with a brush and weigh them.
EXPRESSION OF RESULTS
7. Percentage of material passing sieve of 0.125 mm apertures = (50 − M1) × 2
Percentage of material passing sieve of 0.063 mm apertures = [50 − (M1 + M2)] × 2
M1 = weight in g of the residue on the 0.125 mm sieve
M2 = weight in g of the residue on the 0.063 mm sieve.
The results are to be rounded up to the nearest unit.
REMARK
8. If the presence of lumps is observed after sieving the analysis should be carried out again in the following way:
slowly pour 50 g of the sample into a 1 litre flask containing 500 ml of the sodium hexametaphosphate solution, stirring continuously. Stopper the flask and shake vigorously by hand to break up the lumps. Transfer the whole suspension into the top sieve and wash the flask thoroughly. Continue the analysis as described under paragraph 6.
16.METHODS OF ANALYSIS AND TEST PROCEDURES FOR AMMONIUM NITRATE FERTILISERS CONTAINING MORE THAN 28% NITROGEN BY WEIGHT
A.Methods for the Application of Thermal Cycles
SCOPE AND FIELD OF APPLICATION
1. This method defines the procedures for the application of thermal cycles prior to the execution of the oil retention test in straight ammonium nitrate fertilisers containing more than 28% nitrogen by weight.
THERMAL CYCLES
Field application
2.—2.1 This procedure is for thermal cycling prior to determining the oil retention of the fertiliser.
Principle and definition
2.2 In an Erlenmeyer flask, heat the sample from ambient temperature to 50°C and maintain at this temperature for a period of two hours (phase at 50°C). Thereupon, cool the sample until a temperature of 25°C is acheived and maintain at that temperature for two hours (phase at 25°C). The combination of the successive phases at 50°C and 25°C forms one thermal cycle. After being subjected to two thermal cycles, the test sample is held at a temperature of 20±3°C for the determination of the oil retention value.
Apparatus
2.3 Normal laboratory apparatus, in particular
Water baths thermostated at 25 (±1) and 50 (±1)°C respectively.
Erlenmeyer flasks with an individual capacity of 150 ml.
Procedure
2.4 Put each test sample of 70(±5) grams into an Erlenmeyer flask which is then sealed with a stopper.
Move each flask every two hours from the 50°C bath to the 25°C bath and vice versa.
Maintain the water in each bath at constant temperature and keep in motion by rapid stirring to ensure the water level comes above the level of the sample. Protect the stopper from condensation by a foam rubber cap.
B.Determination of Oil Retention
SCOPE AND FIELD OF APPLICATION
1. This method defines the procedure for the determination of oil retention of straight ammonium nitrate fertilisers containing more than 28% nitrogen by weight.
The method is applicable to both prilled and granular fertilisers which do not contain oil-soluble materials.
DEFINITION
2. Oil retention of a fertiliser: the quantity of oil retained by the fertiliser determined under the operating conditions specified and expressed as a percentage by mass.
PRINCIPLE
3. Total immersion of the test portion in gas oil for a specified period, followed by the draining away of surplus oil under specified conditions. Measurement of the increase in mass of the test portion.
REAGENT
4.
| Gas oil | |
| Viscosity max: | 5 mPas at 40°C |
| Density: | 0.8 to 0.85 g/ml at 20°C |
| Sulphur content: | ≤1.0% (m/m) |
| Ash: | ≤0.1% (m/m) |
APPARATUS
5. Ordinary laboratory apparatus and:
5.1 Balance, capable of weighing to the nearest 0.01 gram.
5.2 Beakers, of capacity 500 ml.
5.3 Funnel, of plastic materials, preferably with a cylindrical wall at the upper end, diameter approximately 200 mm.
5.4 Test sieve, aperture 0.5 mm, fitting into the funnel (5.3).
Note The size of the funnel and sieve is such as to ensure that only a few granules lie one above another and the oil is able to drain away.
5.5 Filter paper, rapid filtering grade, creped, soft, weight 150g/m2.
5.6 Absorbent tissue (laboratory grade).
PROCEDURE
6.—6.1 Two individual determinations are carried out in quick succession on seperate portions of the same test sample.
6.2 Remove particles smaller than 0.5 mm using the test sieve (5.4). Weigh to the nearest 0.01 gram approximately 50 grams of the sample into the beaker (5.2). Add sufficient gas oil (Section 4) to cover the prills completely and stir carefully to ensure that the surfaces of all the prills are fully wetted. Cover the beaker with a watch galss and leave to stand for one hour at 25(±2)°C.
6.3 Filter the entire contents of the beaker through the funnel (5.3) containing the test sieve (5.4). Allow the portion retained by the sieve to remain there for one hour so that most of the excess oil can drain away.
6.4 Lay two sheets of filter paper (5.5) (about 500 × 500 mm) on top of each other on a smooth surface; fold the four edges of both filter papers upwards to a width of about 40 mm to prevent the prills from rolling away. Place two layers of absorbent tissue (5.6) in the centre of the filter papers. Pour the entire contents of the sieve (5.4) over the absorbent tissues and spread the prills evenly with a sof flat brush. After two minutes lift one side of the tissues to transfer the prills to the filter papers beneath and spread them evenly over these with the brush. Lay another sheet of filter paper, similarly with its edges turned upward, on the sample and roll the prills between the filter papers with circular movements while exerting a little pressure. Pause after every eight circular movements to lift the opposite edges of the filter papers and return to the centre the prills that have rolled to the periphery. Keep to the following procedure: make four complete circular movements first clockwise and then anticlockwise. Then roll the prills back to the centre as described above. This procedure to be carried out three times (24 circular movements, edges lifted twice). Carefully insert a new sheet of filter paper between the bottom sheet and the one above it and allow the prills to roll onto the new sheet by lifting the edges of the upper sheet. Cover the prills with a new sheet of filter paper and repeat the same procedure as described above. Immediately after rolling, pour the prills into a tared dish and reweigh to the nearest 0.01 gram to determine the weight of the quantity of gas oil retained.
Repeating the rolling procedure and reweighing
6.5 If the quantity of gas oil retained in the portion is found to be greater than 2.00 grams, place the portion on a fresh set of filter papers and repeat the rolling procedure, lifting the corners in accordance with Section 6.3 (two times eight circular movements, lifting once). Then reweigh the portion.
EXPRESSION OF RESULTS
Method of calculation and formula
7.—7.1 The oil retention, from each determination (6.1) expressed as a percentage by mass of the sieved test portion, is given by the equation:

where:
m1 is the mass, in grams, of the sieved test portion (6.2);
m2 is the mass, in grmas, of the test portion accodring to Section 6.4 or 6.5 respectively as the result of the last weighing.
Take as the result the arithmetic mean of the two individual determinations.
C.Determination of the Combustible Ingredients
SCOPE AND FIELD OF APPLICATION
1. This method defines the procedure for the determination of the combustible content of straight ammonium nitrate fertilisers containing more than 28% nitrogen by weight.
PRINCIPLE
2. The carbon dioxide produced by inorganic filters is removed in advance with an acid. The organic compounds are oxidised by means of a chromic acid/sulphuric acid mixture. Carbon dioxide formed is absorbed in a barium hydroxide solution. The precipitate is dissolved in a solution of hydrochloric acid and measured by back-titration with sodium hydroxide solution.
REAGENTS
3.—3.1 Analytical-grade chromium VI oxide; Cr-(VI)-)3.
3.2 Sulphuric acid diluted to 60% by volume:
pour 360 ml of water into a one-litre beaker and carefully add 640 ml of sulphuric acid, density at 20°C=1.83 g/ml.
3.3 Silver nitrate: 0.1 M solution.
3.4 Barium hydroxide:
weigh out 15 grams of barium hydroxide (Ba(OH)2.8H2O), and dissolve completely in hot water.
Allow to cool and transfer to a one-litre flask. Fill up to the mark and mix. Filter through a pleated filter paper.
3.5 Hydrochloric acid : 0.1 M standard solution.
3.6 Sodium hydroxide: 0.1 M standard solution.
3.7 Bromophenol blue: solution of 0.4 grams per litre in water.
3.8 Phenolphthalein: solution of 2 grams per litre in 60% by volume ethanol.
3.9 Soda lime: particle dimensions, about 1.0 to 1.5 mm.
3.10 Demineralised water, freshly boiled to remove carbon dioxide.
APPARATUS
4.—4.1 Standard laboratory equipment, in particular:
filter crucible with a plate of sintered glass and a capacity of 15 ml, plate diameter: 20 mm, total height : 50 mm, porosity 4 (pore diameter from 5 to 15μm);
600 ml beaker.
4.2 Compressed nitrogen supply.
4.3 Apparatus made up of the following parts and assembled, if possible, by means of spherical ground joints (see Figure 1).
4.3.1Absorption tube (A) about 200 mm long and 30 mm in diameter filled with soda lime (3.9) kept in place by fibreglass plugs.
4.3.2500 ml reaction flask (B) with side arm and a round bottom.
4.3.3Vigreux fractionating column about 150 mm long (C’).
4.3.4Double-surface condenser (C), 200 mm long.
4.3.5Drechsel bottle (D) acting as a trap for any excess acid which may distil over.
4.3.6Ice bath (E) to cool the Drechsel bottle.
4.3.7Two absorption vessels (F1) and (F2), 32 to 35 mm in diameter, the gas distributor of which comprises a 10 mm disc of low-porosity sintered glass.
4.3.8Suction pump and suction regulating device (G) comprising a T-shaped glass piece inserted into the circuit, the free arm of which is connected to a fine capillary tube by a short rubber tube fitted with a screw clamp.
Caution:
the use of boiling chromic acid solution in an apparatus under reduced pressure is a hazardous operation and requires appropriate precautions.
PROCEDURE
Sample for analysis
5.—5.1 Weigh approximately 10 grams of ammonium nitrate to the nearest 0.001 grams.
Removal of carbonates
5.2 Place the sample for analysis in the reaction flask (B). Add 100 ml of H2So4 (3.2). The prills dissolve in about 10 minutes at ambient temperature. Assemble the apparatus as indicated in the diagram: connect one end of the absorption tube (A) to the nitrogen source (4.2) via a non-return flow device containing 5 to 6 mm of mercury and the other end to the feed tube which enters the reaction flask. Place the Vigreux fractionating column (C’) and the condenser (C) with coolinhg water supply in position. Adjust the nitrogen to provide a moderate flow through the solution, bring the solution to boiling point and heat for two minutes. At the end of this time there should be no more effervesence, if effervesence is seen, continue heating for 30 minutes. Allow solution to cool for at least 20 minutes with the nitrogen flowing through it.
Complete assembly of the apparatus as indicated in the diagram by connecting the condenser tube to the Drechsel bottle (D) and the bottle to the absorption vessels F1 and F2. The nitrogen must continue to pass through the solution during the assembly operation. Rapidly introduce 50 ml of barium hydroxide solution (3.4) into each of the absorption vessels (F1 and F2).
Bubble a stream of nitrogen through for about 10 minutes. The solution must remain clear in the absorbers. If this does not happen, the carbonate removal process must be adjusted.
Oxidation and absorption
5.3 After withdrawing the nitrogen feed tube, rapidly introduce 20 grams of chromium trioxide (3.1) and 6 ml of silver nitrate solution (3.3) via the side arm of the reaction flask (B). Connect the apparatus to the suction pump and adjust the nitrogen flow so that a steady stream of gas bubbles passes through the sintered-glass absorbers (F1) and (F2).
Heat the reaction flask (B) until the liquid boils and keep it boiling for one-and-a-half hours(31). It may be necessary to adjust the suction-regulating valve (G) to control the nitrogen flow since it is possible that the barium carbonate precipitated during the test may block the sintered glass discs. The operation is satisfactory when the barium hydroxide solution in the absorber (F2) remains clear. Otherwise repeat the test. Stop heating and dismantle the apparatus. Wash each of the distributors both inside and outside to remove barium hydroxide and collect the washings in the corresponding absorber. Place the distributors one after the other in a 600 ml beaker which will subsequently be used for the determination.
Rapidly filter under vacuum firstly the contents of absorber F2 and then of absorber F1 using the sintered glass crucible. Collect the precipitate by rinsing the absorbers with water (3.10) and wash the crucible with 50 ml of the same water. Place the crucible in the 600 ml beaker and add about 100 ml of boiled water (3.10). Introduce 50 ml of boiled water into each of the absorbers and pass nitrogen through the distributors for five minutes. Combine the water with that fro the beaker. Repeat the operation once to ensure that the distributors are rinsed thoroughly.
Measurement of the carbonates originating from organic material
5.4 Add five drops of phenolphthalein (3.8) to the contents of the beaker. The solution becomes red in colour. Add hydrochloric acid (3.5) drop by drop until the pink colour just disappears. Stir the solution well in the crucible to check that the pink colour does not reappear. Add five drops of bromophenol blue and titrate with hydrochloric acid until the solution turns yellow. Add a further 10 ml of hydrochloric acid.
Heat the solution to boiling point and continue boiling for a maximum of one minute. Check carefully that no precipitate remains in the liquid.
Allow to cool and back titrate with the sodium hydroxide solution (3.6).
BLANK TEST
6. Carry out a blank test following the same procedure and using the same quantities of all reagents.
EXPRESSION OF RESULTS
7. The content of combustible ingredients (C), expressed as carbon, as a percentage by mass of the sample, is given by the formula:

where:
E = the mass in grams of the test portion;
V1 = the total volume in ml of 0.1 M hydrochloric acid added after the change in colour of the phenolphthalein;
V2 = the volume in ml of the 0.1 M sodium hydroxide solution used for back titration.
D.Determination of the pH value
SCOPE AND FIELD OF APPLICATION
1. This method defines the procedure for measuring the pH value of a solution of a straight ammonium nitrate fertiliser containing more than 28% nitrogen by weight.
PRINCIPLE
2. Measurement of the pH of an ammonium nitrate solution by means of a pH meter.
REAGENTS
3. Distilled or demineralised water, free from carbon dioxide.
Buffer solution, pH 6.88 at 20°C
3.1 Dissolve 3.40±0.01 grams of potassium dihydrogen orthophosphate (KH2PO4) in approximately 400 ml of water. The dissolve 3.55±0.01 gram of disodium hydrogen orthophosphate (Na2HPO4) in approximately 400 ml of water. Transfer the two solutions without loss into a 1,00 ml standard flask, make up to the mark and mix. Keep the solution in an airtight vessel.
Buffer solution, pH 4.00 at 20°C
3.2 Dissolve 10.21±0.01 grams of potassium hydrogen phthalate (KHC8O4H4) in water, transfer without loss into a 1,00 ml standard flask, make up to the mark and mix.
Keep this solution in an airtight vessel.
3.3 Commercially available pH standard solutions may be used.
APPARATUS
4. pH meter, equipped with glass and calomel electrodes or equivalent, sensitivity 0.05 pH unit.
APPARATUS
Calibration of the pH meter
5.—5.1 Calibrate the pH meter (4) at a temperature of 20(±1)°C, using the buffer solutions (3.1), (3.2) or (3.3). Pass a slow stream of nitrogen onto the surface of the solution and maintain this throughout the test.
Determination
5.2 Pour 100.0 ml of water onto 10 (±0.01) grams of the sample in a 250 ml beaker. Remove the insolubles by filtering, decanting or centrifuging the liquid.
Measure the pH value of the clear solution at a temperature of 20 (±1)°C according to the same procedure as for the calibration of the meter.
EXPRESSION OF RESULTS
6. Express the result in pH units, to the nearest 0.1 unit and state the temperature used.
E.Determination of the Particle Size
SCOPE AND FIELD OF APPLICATION
1. This method defines the procedure for the test sieving of straight ammonium nitrate fertilisers containing more than 28% nitrogen by weight.
PRINCIPLE
2. The test sample is sieved on a nest of three sieves, either by hand or by mechanical means. The mass retained on each sieve is recorded and the percentage of material passing the required sieves are calculated.
APPARATUS
3.—3.1 200 mm diameter woven-wire test sieves to BS 410 (1986) with apertures of 2.0 mm, 1.0 mm and 0.5 mm respectively of standard ranges. One lid and one receiver for these sieves.
3.2 Balance to weigh to 0.1 gram.
3.3 Mechanical sieve shaker (if available) capable of imparting both vertical and horizontal motion to the test sample.
PROCEDURE
4.—4.1 The sample is divided representatively into portions of approximately 100 grams.
4.2 Weigh one of these portions to the nearest 0.1 gram.
4.3 Arrange the nest of sieves in ascending order; receiver 0.5 mm, 1 mm, 2 mm and place the weighed test portion on the top sieve. Fit the lid to the top of the nest of sieves.
4.4 Shake by hand or machine, imparting both a vertical and horizontal motion and, if by hand, tapping occasionally. Continue this process for 10 minutes or until the quantity passing through each sieve in one minute is less than 0.1 gram.
4.5 Remove the sieves from the nest in turn and collect the material retained, brush gently from the reverse side with a soft brush, if necessary.
4.6 Weigh the material retained on each sieve and that collected in the receiver, to the nearest 0.1 gram.
EVALUATION OF RESULTS
5.—5.1 Convert the fraction masses to a percentage of the total of the fraction masses (not of the original charge).
Calculate the percentage in the receiver (ie <0.5 mm): A%
Calculate the percentage retained on the 0.5 mm sieve: B%
Calculate the percentage passing 1.0 mm, ie (A + B)%.
The sum of the fraction masses should be within 2% of the initial mass taken.
5.2 At least two seperate analyses should be carried out and the individual results for A should not differ by more than 1.0% absolute and for B by more than 1.5% absolute. Repeat the test if this is not the case.
EXPRESSION OF RESULTS
6. Report the mean of the two values for A on the one hand and for A + B on the other hand.
F.Determination of the Chlorine Content (as Chloride Ion)
SCOPE AND FIELD OF APPLICATION
1. This method defines the procedure for the determination of the chlorine content (as chloride ion) of straight ammonium nitrate fertilisers containing more than 28% nitrogen by weight.
PRINCIPLE
2. Chloride ions dissolved in water are determined by potentiometric titration with silver nitrate in an acidic medium.
REAGENTS
3. Distilled or demineralised water, free from chloride ions.
3.1 Acetone AR.
3.2 Concentrated nitric acid (density at 20°C = 1.40 g/ml).
3.3 Silver nitrate 0.1 M standard solution. Store this solution in a brown glass bottle.
3.4 Silver nitrate 0.004 M standard solution—prepare this solution at the time of use.
3.5 Potassium chloride 0.1 M standard reference solution. Weigh, to the nearest 0.1 mg, 3.7276 grams of analytical-grade potassium chloride, previously dried for one hour in an oven at 130°C and cooled in a desiccator to ambient temperature. Dissolve in a little water, transfer the solution without loss into a 500 ml standard flask, dilute to the mark and mix.
3.6 Potassium chloride 0.004 M standard reference solution—prepare this solution at the time of use.
APPARATUS
4.—4.1 Potentiometer with silver indicating electrode and calomel reference electrode, sensitivity 2 mV, covering the range −500 to +500 mV, or with silver and mercury (1) sulphate electrodes.
4.2 Bridge, containing a saturated potassium nitrate solution, connected to the calomel electrode (4.1), fitted at the ends with porous plugs. This bridge is not necessary if silver and mercury (1) sulphate electrodes are used.
4.3 Magnetic stirrer, with a Teflon-coated rod.
4.4 Microburette with a fine-pointed tip, graduated in 0.01 ml divisions.
PROCEDURE
Standardisation of the silver nitrate solution
5.—5.1 Take 5.00 ml and 10.00 ml of the standard reference potassium chloride solution (3.6) and place in two low-form beakers of convenient capacity (for example 250 ml). Carry out the following titration of the contents of each beaker.
Add 5 ml of the nitric acid solution (3.2), 120 ml of the acetone (3.1) and sufficient water to bring the total volume to about 150 ml. Place the rod of the magnetic stirrer (4.3) in the beaker and set the stirrer in motion. Immerse the silver electrode (4.1) and the free end of the bridge (4.2) in the solution. Connect the elctrodes to the potentiometer (4.1) and, after verifying the zero of the apparatus, note the value of the starting potential.
Titrate, using the microburette (4.4), adding initially 4 or 9 ml respectively of the silver nitrate solution corresponding to the standard reference potassium chloride solution used. Continue the addition in 0.1 ml portions for the 0.004 M solutions and in 0.05 ml portions for the 0.1 M solutions. After each addition, await the stabilisation of the potential.
Record the volumes added and the corresponding values of the potential in the first two columns of a table.
In a third column of the table, record the successive increments (Δ1E) of the potential E. In a fourth column, record the differences (Δ2E) positive or negative, between the potential increments (Δ1E). The end of the titration corresponds to the addition of the 0.1 or 0.05 ml portion (V1) of the silver nitrate solution which gives the maximum value of (Δ1E).
In order to calculate the exact volume (Veq) of the silver nitrate solution corresponding to the end of the reaction, use the formula:
where:
V0 is the total volume, in ml, of the silver nitrate solution immediately lower than the volume which gives the maximum increment of Δ1E;
V1 is the volume, in ml, of the last portion of the silver nitrate solution added (0.1 or 0.05 ml);
b is the last positive value of Δ2E;
B is the sum of the absolute values of the last positive values of Δ2E and the first negative value of Δ2E (see example in Table 1).

Blank test
5.2 Carry out a blank test and take account thereof when calculating the final result.
The result V4 of the blank test and take account thereof when calculating the final result.
V4 = 2V3 - V2
where:
V2 is the value, in ml, of the exact volume (Veq) of the silver nitrate solution corresponding to the titration of 10 ml of the potassium chloride standard reference solution used;
V3 is the value, in ml, of the exact volume (Veq) of the silver nitrate solution corresponding to the titration of 5 ml of the potassium chloride standard reference solution used.
Check test
5.3 The blank test can at the same time serve as a check that the apparatus is functioning satisfactorily and that the test procedure is being implemented correctly.
Determination
5.4 Take a portion of sample in the range of 10 to 20 grams and weigh to the nearest 0.01 gram. Transfer quantitatively to a 250 ml beaker. Add 20 ml of water, 5 ml of nitric acid solution (3.2), 120 ml of acetone (3. 1) and sufficient water to bring the total volume to about 150 ml.
Place the rod of the magnetic stirrer (4.3) in the beaker, place the beaker on the stirrer and set the stirrer in motion. Immerse the silver electrode (4.1) and the free end of the bridge (4.2) in the solution, connect the electrodes to the potentiometler (4. 1) and, after having verified the zero of the apparatus, note the value of the starting potential.
Titrate with the silver nitrate solution, by additions from the microburette (4.4) in increments of 0.1 ml. After each addition, await the stabilisation of the potential.
Continue the titration as specified in 5. 1, starting from the fourth paragraph: “Record the volumes added and the corresponding values of the potential in the first two columns of a table….”.
EXPRESSION OF RESULTS
6. Express the result of the analysis as the percentage of chlorine contained in the sample as received for analysis.
Calculate the percentage of chlorine (CI) content from the formula:

where:
T is the molarity of silver nitrate solution used;
V4 is the result, in ml, of the blank test (5.2);
V5 is the value, in ml, of Veq corresponding to the determination (5.4);
m is the mass, in grams, of the test portion.
Table 1
EXAMPLE
| Volume of the silver nitrate solution V | Potential E | ||
|---|---|---|---|
| ml | mv | Δ1E | Δ2E |
| 4.80 | 176 | ||
| 4.90 | 211 | 72 | 35 |
| 5.00 | 283 | 72 | +37 |
| 5.10 | 306 | 23 | −49 |
| 5.20 | 319 | 13 | −10 |

G.Determination of Copper
SCOPE AND FIELD OF APPLICATION
1. This method defines the procedure for the determination of copper content of straight ammonium nitrate fertilisers containing more than 28% nitrogen by weight.
PRINCIPLE
2. The sample is dissolved in dilute hydrochloric acid and the copper content is determined by atomic absorption spectrophotometry.
REAGENTS
3.—3.1 Hydrochloric acid (density at 20°C=1.18 g/ml).
3.2 Hydrochloric acid, 6 M solution.
3.3 Hydrochloric acid, 0.5 M solution.
3.4 Ammonium nitrate.
3.5 Hydrogen peroxide, 30%
3.6 Copper solution(32) (stock): weigh, to the nearest 0.001 gram, 1 gram of pure copper, dissolve in 25 ml 6 M hydrochloric acid solution (3.2), add 5 ml of hydrogen peroxide (3.5) in portions and dilute to I litre with water. 1 ml of this solution contains 1,000,μg of copper (Cu).
3.6.1Copper solution (dilute): dilute 10 ml of stock solution (3.6) to 100 ml with water and then dilute 10 ml of the resulting solution to 100 ml with water. 1 ml of the final dilution contains 10μg of copper (Cu).
Prepare this solution at the time of use.
APPARATUS
4. Atomic absorption spectrophotometer with a copper lamp (324.8 nm).
PROCEDURE
Preparation of the solution for analysis
5.—5.1 Weigh, to the nearest O.OO1 gram, 25 grams of the sample, place it in a 400 ml beaker, add carefully 20 ml of hydrochloric acid (3.1) (there may be a vigorous reaction due to carbon dioxide formation). Add more hydrochloric acid, if necessary. When effervescence has stopped, evaporate to dryness on a steam bath, stirring occasionally with a glass rod. Add 15 ml 6 M hydrochloric acid solution (3.2) and 120 ml of water. Stir with the glass rod, which should be left in the beaker and cover the beaker with a watch glass. Boil the solution gently until dissolution is complete and then cool.
Transfer the solution quantitatively into a 250 ml graduated flask, by washing the beaker with 5 ml 6 M hydrochloric acid (3.2), and twice with 5 ml of boiling water, make up to the mark with 0.5 M hydrochloric acid (3.3) and mix carefully.
Filter through a copper-free filter paper(33), discarding the first 50 ml.
Blank solution
5.2 Prepare a blank solution from which only the sample has been omitted and allow for this in the calculation of the final result.
Determination
5.3.1 Preparation of sample and blank test solutions
5.3.1Dilute the sample solution (5.1) and the blank test solution (5.2) with 0.5 M hydrochloric acid solution (3.3) to a concentration of copper within the optimal measuring range of the spectrophotometer. Normally no dilution is needed.
5.3.2 Preparation of the calibration solutions
5.3.2By diluting the standard solution (3.6.1) with 0.5 M hydrochloric acid solution (3.3), prepare at least five standard solutions corresponding to the optimal measuring range of the spectrophotometer (0 to 5.0 μ/l Cu). Before making up to the mark, add to every solution ammonium nitrate (3.4) to give a final concentration of 100 mg per ml.
Measurement
5.4 Set up the spectrophotometer (4) at a wavelength of 324.8 nm using an oxidising air-acetylene flame. Spray successively, in triplicate, the calibration solutions (5.3.2), the sample solution and the blank solution (5.3.1), washing the instrument through with distilled water between each spraying. Plot the calibration curve using the mean absorbances of every standard used as the ordinates and the corresponding concentrations of copper in μ/ml as the abscissae.
Determine the concentration of copper in the final sample and blank solutions by reference to the calibration curve.
EXPRESSION OF RESULTS
6. Calculate the copper content of the sample taking into account the weight of the test sample, the dilutions carried out in the course of the analysis and the value of the blank. Express the result as mg Cu/kg.
PART II
General
1.—(a) When two or more methods are prescribed in this part of this Schedule to determine a component of a fertiliser the choice of the method shall, except where otherwise indicated, be left to the agricultural analyst concerned; the method used must however be indicated in the certificate of the analysis.
(b)Any reference to water in this Schedule means purified water as defined in the European Pharmacopeia.
Reagents and Apparatus
2.—(a) All reagents used shall be of analytical quality.
(b)For the determination of any form of nitrogen, water must be free of all nitrogeneous compounds and carbon dioxide.
(c)Solutions for which no solvents are prescribed must be aqueous.
(d)Only special instruments or apparatus requiring special standards are mentioned in the descriptions of the methods of analysis.
Methods of Analysis
1. Preparation of the sample for analysis
2. Determination of moisture
3. Determination of total nitrogen—chromium powder reduction method
4. Determination of urea
5.a Extraction of phosphorus—by mineral acids (total phosphorus)
b Extraction of phosphorus—by 2% citric acid
6. Determination of extracted phosphorus—spectrophotometric method
7.a Determination of potassium—gravimetric method
b Determination of potassium—flame photometric method
8. Determination of total magnesium
9.a. Determination of boron—titrimetric method
b Determination of boron—spectrophotometric method
10. Determination of cobalt
11. Determination of molybdenum
12. Determination of copper
13. Determination of iron
14. Determination of manganese
15. Determination of the nuetralising value in limiting materials
16. Determination of fineness of products other than potassic bag slag
17. Determination of fineness of potassic basic slag.
1.PREPARATION OF THE SAMPLE FOR ANALYSIS
INTRODUCTION
1. The preparation of a sample for analysis from the final sample received at the laboratory is a series of operations, usually sieving, grinding and mixing carried out in such a way that the smallest amount weighed, as prescribed by the method of analysis chosen, is representative of the final sample. The sample should be ground to the fineness required by the method of analysis. (Overgrinding must be avoided in cases where this will affect the solubility in various reagents.) With some materials, fine grinding may lead to loss of gain or moisture and allowance for this must be made.
SCOPE AND FIELD OF APPLICATION
2. This method is applicable to fertilisers in Groups 1(b), 1(c), 2(d), 3(b), 3(c), 4(a), 4(b), 4(c) of Section A and Group 5 of Section B of the Table in Schedule 1 of the Fertilisers Regulations 1990(34). It is also applicable to products in Group 5(a) of Section A in the said table when the determination of total magnesium is required.
This method is also applicable to fluid fertilisers.
The determination of the fineness of fertilisers is carried out on the sample as received.
PRINCIPLE
3.—3.1 Solid fertilisers: the whole final sample is ground to the required fineness. All the ground sample is thoroughly mixed before each test portion is taken.
3.2 Fluid fertilisers: the final sample is thoroughly mixed before each test portion is taken.
APPARATUS
4.—4.1 Sample grinder capable of grinding the fertiliser to pass the specified sieve.
4.2 Mortar and pestle of suitable material and size.
4.3 Sieves having square apertures of 0.18 mm, 0.5 mm and 1.0 mm. Test sieves conforming to British Standard 410; 1976 are suitable.
4.4 Sample containers of non-corrodible materials, with air tight closures.
PROCEDURE
WARNING
All operations connected with this procedure should be carried out as quickly as possible to minimise absorbtion or loss of water. Care should be taken during grinding that the temperature of the fertiliser does not rise above 45°C to avoid loss of volatile constituents. Grinding beyond the fineness required must in all cases be avoided.
Grinding and sieving
5.1 The procedure in 5.1.1 should be followed except when a grinding machine is not available, in which case 5.1.2 is applicable.
5.1.1Grind the final sample until all the sample has passed through, or for the specified time, depending on the type of grinder (4.1). To check that the grinding has been adequate sieve a small portion of the ground sample through a 0.5 mm sieve (4.3) and discard it. If the whole of this portion does not pass the sieve, return the remainder of the sample to the grinder and repeat the grinding until satisfactory grinding is achieved.
5.1.2Sieve the whole final sample through a 0.5 mm sieve (4.3). Grind the residue on the sieve, using the pestle and mortar (4.2), until all the material passes through the sieve. Carefully mix the sample.
5.2 Place the prepared sample in a clean container (4.4) and seal it until required for analysis.
5.3 Before taking each test portion for analysis, the whole sample must be well mixed. Form the material into a flattened cone and using a spatula take the required test portion at random in small increments.
5.4 If the sample contains foreign matter which cannot be ground this shall be removed, weighed and allowed for in the results of the analysis. This material shall be retained and if possible its nature recorded.
SPECIAL CASES
Samples not to be ground
6.—6.1 For those samples where the amount of phosphorous pentoxide solubile in 2percnt; citric acid and the fineness of grinding are to be determined, the sample should be well mixed (soft lumps may be disintergrated by lightly crushing) and divided into two parts, which are as identical as possible. The above mentioned determinations shall be carried out on the unground sample. All other determinations shall be carried out on the sample prepared in accordance with the directions in paragraph 5.1.
Products which may be difficult to grind mechanically, including products with abnormal moisture or products which become doughy through grinding
6.2 Some products such as superphosphate may become doughy if ground mechanically. In these case crush the sample in a mortar (4.2) so that all the material passes through a 1.0 mm sieve (4.3). Place the material so crushed in a clean container (4.4) and seal it until required for analysis.
Organic materials
6.3 Some organic materials may be of such a nature that the procedures given above cannot be used (for example fresh guano, leather, wool and animal residues). In these cases the analyst should use the best practicable means to obtain a representative sample.
Fertilisers comprising several different materials
6.4 These fertilisers include materials with marked differences in texture or mechanical properties (hardness, density, etc). They may be difficult to grind entirely (for example mixtures of organic and inorganic materials) or they may segregate during handling (for example “Kalimagnesia”). Special procedures are necessary in these cases:
6.4.1for mixtures other than those in 6.4.2, follow the procedure in 5.1.1, repacing the 0.5 mm sieve by one with apertures of 0.18 mm. A grinding machine, capable of grinding the whole of the sample to the required fineness in one pass, is strongly recommended;
6.4.2in the case of mixtures containing one or more very hard components, or mixtures containing organic materials, it may be difficult to grind and homogenise all the components. To avoid overgrinding some of the softer components proceed as follows:—
grind the sample as in 5.1.1 or 5.1.2 to pass a 0.5 mm sieve. Re-seive the sample through a 0.18 mm sieve and reduce the residue to a convenient size by further grinding or other practicable means. Thoroughly remix the sample and place in a clean container (4.4).
FLUID FERTILISERS
7. Mix thoroughly by shaking, ensuring that any insoluble matter, particularly crystaline material, is throughly dispersed, immediately before drawing a portion of the sample for analysis.
2.DETERMINATION OF MOISTURE
SCOPE AND FIELD OF APLICATION
1. This method is applicable to all fertilisers where a correction for moisture is necessary.
PRINCIPLE
2. The sample is dried to constant weight in an oven at 100°C. The loss in weight corresponds to the moisture content of the sample.
APPARATUS
3.—3.1 Suitable containers with lids ensuring air tight closure; the dimensions should allow the sample to be spread at about 0.3 g per cm2.
3.2 Electrically heated oven, suitably ventilated and capable of being maintained at 100±2°C.
PREPARATION OF SAMPLE
4. See Method 1.
PROCEDURE
5. Weigh to the nearest 0.001 g, 5 g of the prepared sample and transfer to a previously weighed container (3.1). Place the uncovered container and the lid in the oven (3.2) for 2 to 3 hours. Replace the lid on the container, remove from the oven and allow to cool in a desiccator and weigh. Reheat for another hour, cool and reweigh. If the difference in weight exceeds 0.01g continue the heating and cooling procedure until a weight constant within 0.01 g is attained.
EXPRESSION OF RESULT
6. Calculate the total loss of weights and express it as a percentage of the original weight.
3.DETERMINATION OF TOTAL NITROGEN-CHROMIUM POWDER REDUCTION METHOD
SCOPE AND FIELD OF APPLICATION
1. This method is applicable to fertilisers in Groups 1(b), 1(c), 3(b), 4(a) and 4(c) of Section A, Group 5 of Section B and Groups 1(c) and 1(d) of Section C of the Table in Schedule 1 of the Fertilisers Regulations 1990(35) in respect of which the indication of total nitrogen is required.
PRINCIPLE
2. The nitrate is reduced to ammonia by chromium powder in an acid medium. Organic and ureic nitrogen is converted into ammonium sulphate by digestion with concentrated sulphuric acid using a catalyst. The ammonia is distilled from an alkaline solution and absorbed in a standard acid. The excess acid is tirated with standard alkali.
REAGENTS
3.—3.1 Sodium hydroxide solution: 40 g per 100 ml, ammonia free.
3.2 Sulphuric acid, 0.1 N solution.
3.3 Sulphuric acid, 0.2 N solution.
3.4 Sulphuric acid, 0.5 N solution.
3.5 Sodium hydroxide, 0.2 N solution, carbonate free.
3.6 Chronium metal powder, 100 mesh, low nitrogen content.
3.7 Anti-bump granules of pumice stone, washed in hydrochloric acid and ignited.
3.8 Anti-foaming agent, paraffin wax.
3.9 Sulphuric acid (d=1.84 g/ml).
3.10 Hydrochloric acid (d=1.18 g/ml).
3.11 Catalyst mixture: 1,000 g potassium sulphate and 50 g copper sulphate pentahydrate. The ingredients must be ground and thoroughly mixed.
3.12 Indicator solutions:
3.12.1 Mixed indicator:
3.12.1mix 50 ml of 2 g/litre ethanolic solution of methyl red with 50 ml of 1 g/litre ethanolic solution of methylene blue.
3.12.2 Methyl red indicator:
3.12.2dissolve 0.1 g methyl red in 50 ml ethanol. This indicator may be used instead of the preceding one.
3.13pH indicator paper, wide range.
APPARATUS
4. Apparatus for mineral acid digestion and distillation according to Kjeldahl’s method.
PREPARATION OF SAMPLE
5. See Method 1.
PROCEDURE
Reduction
6.—6.1 Weigh, to the nearest 0.001 g, between 0.5 and 2.0 g of the prepared sample, containing not more than 0.06 g nitric nitrogen and 0.235 g total nitrogen and transfer to a Kjeldahl flask. Add sufficient water to make the total volume 35 ml. Allow the flask to stand for 10 minutes with occasional gentle swirling to ensure solution of all nitrate salts.
Add 1.2 g chromium powder (3.6) and 7 ml hydrochloric acid (3.10), mix well and allow the flask to stand for at least 5 minutes but not more than 10 minutes at ambient temperature. Heat the flask gently so that the contents just begin to boil in about 7 minutes. Continue boiling gently for 10 minutes. Remove the flask from the heat and allow to cool.
Hydrolysis, when the fertiliser is known not to contain organic matter
6.2 Place the flask (6.1) in a fume cupboard, add a small quantity of anti-bump granules (3.7) and then carefully add 25 ml sulphuric acid (3.9). Mix the contents of the flask and heat gently until boiling. Continue heating until dense white fumes of sulphuric acid are evolved for at least 15 minutes. Allow the mixture to cool and then carefully add 250 ml water. Allow to cool to room temperature and continue as described in 6.4.
Digestion when the fertiliser is known to contain organic matter
6.3 Add a small quantity of anti-bump granules (3.7), 10 g of the catalyst mixture (3.11) and then carefully add 25 ml sulphuric acid (3.9) (see Note). Add 0.5 g parafin wax (3.8) to reduce foaming and mix. Heat the flask moderately at first, shaking from time to time until frothing ceases and the liquid is practically colourless. Continue the digestion for at least a further 60 minutes. Allow the mixture to cool and then carefully add 250 ml water. Allow to cool to room temperature, and continue as described in 6.4.
Note: If organic matter other than urea exceeds 1.0 g add an additional 1.0 ml sulphuric acid for each 0.1 g organic matter in excess of 1.0 g.
Distillation
6.4 Transfer an appropriate volume of 0.1 N, 0.2 N or 0.5 N sulphuric acid (3.2, 3.3, 3.4) to the collecting flask of the distillation apparatus, according to the presumed level of nitrogen; add a few drops of indicator solution (3.12.1 or 3.12.2). Taking precautions against the loss of ammonia, carefully add to the contents of the Kjeldahl flask (6.2 or 6.3) 100 ml sodium hydroxide solution (3.1). Mix well and connect immediately to the distillation apparatus. Heat the flask so that approximately 150 ml of the liquid are distilled in 30 minutes. At the end of this time, lower the collecting flask so that the tip of the condenser is above the surface of the liquid. Test the subsequent distilate by means of the indicator paper (3.13) to ensure that all the ammonia is completely distilled. Remove the source of heat. Titrate the excess acid with 0.2 N sodium hydroxide solution (3.5) to the end point of the indicator.
Blank test
6.5 Carry out a blank test (omitting only the sample) under the same conditions and allow for this in the calculation of the final results.
EXPRESSION OF RESULTS
7. Determine the quantity of sulphuric acid consumed.
1 ml 0.1 N sulphuric acid=0.0014 g nitrogen.
1 ml 0.2 N sulphuric acid=0.0028 g nitrogen.
1 ml 0.5 N sulphuric acid=0.0070 g nitrogen.
Express the result as the percentage of nitrogen (N) contained in the fertiliser as received for analysis.
4.DETERMINATION OF UREA
SCOPE AND FIELD OF APPLICATION
1. This method is applicable to fertilisers in Group 1(c) of Section A, Group 5 of Section B and Group 1(d) of Section C of the Table in Schedule 1 of the Fertilisers Regulations 1990(36).
PRINCIPLE
2. The sample is suspended in acid solution with a clarifying agent and filtered. The urea content of the filtrate is determined after the addition of 4-dimethylamino-benzaldehyde (4-DMAB) by measuring the absorbance at 435 nm.
REAGENTS
3.—3.1 Activated charcoal.
3.2 Carrez solution I:
dissolve 21.9 g zinc acetate dihydrate in water, add 3 ml glacial acetic acid and dilute to 100 ml with water.
3.3 Carrez solution II: 10.6 g potassium ferrocyanide per 100 ml.
3.4 Hydrochloric acid solution 0.02 N.
3.5 Sodium acetate solution: 136 g sodium acetate trihydrate per litre.
3.6 4-dimethylamino-benzaldehyde solution:
dissolve 1.6 g of 4-dimethylamino-benzaldehyde (4-DMAB) in 100 ml 96percnt; ethanol and add 10 ml of hydrochloric acid (d=1.18 g/ml).
3.7 Urea standard solution: 1.0 g per 100 ml (1 ml of this solution=10 mg urea).
APPARATUS
4. Spectrophotometer with 10 mm cells.
PREPARATION OF SAMPLE
5. See Method 1.
PROCEDURE
Preparation of the solution for analysis
6.—6.1 Weigh to the nearest 0.001 g, 2 g of the prepared sample, or a suitable amount expected to contain between 50 and 500 mg of urea and transfer it to a 500 ml graduated flask. Add 150 ml 0.02 N hydrochloric acid solution (3.4), shake for 30 minutes then add 10 ml sodium acetate solution (3.5) and mix well. Add 2 g activated charcol (3.1) to the flask, shake well and allow to stand for a further 15 minutes. Add 5 ml Carrez solution I (3.2), followed by 5 ml Carrez solution II (3.3), mixing well between additions. Dilute to volume with water and mix well. Filter a portion of the solution through a dry filter paper into a clean dry filter paper into a clean dry 250 ml beaker.
Determination
6.2 Transfer 10 ml of the filtrate (6.1) to a 50 ml graduated flask, add 10 ml 4-DMAB solution (3.6), dilute to 50 ml with water mix well and allow to stand for 10 minutes. Measure the absorbance of the solution at 435 nm, in a 10 mm cell against a reference solution prepared by diluting 10 ml 4-DMAB solution (3.6) to 50 ml with water.
Calibration curve
6.3 Transfer amounts of standard urea solution (3.7) corresponding to 50, 100, 150 and 250 mg of urea into a series of 250 ml graduated flasks; add 75 ml 0.02 N hydrochloric acid solution (3.4) and proceed as described above (6.1) commencing at “…shake for 30 minutes…”. Measure the absorbance of the solutions and construct a calibration graph relating the absorbances to the amounts of urea present.
EXPRESSION OF RESULTS
7. Determine the amount of urea in the sample by reference to the calibration graph. Express the result in terms of percentage ureic nitrogen of the sample:
(mg urea × 0.4665=mg ureic nitrogen).
5a.EXTRACTION OF PHOSPHORUS BY MINERAL ACIDS (TOTAL PHOSPHOROUS)
SCOPE AND FIELD OF APPLICATION
1. This method is applicable to fertilisers in Groups 2(b), 2(c), 2(d), 3(b) and 4(c) of Section A and Group 5 of Section B of the Table in Schedule 1 of the Fertilisers Regulations 1990(37) in respect of which the indication of total phosphorus is required.
PRINCIPLE
2. The phosphorous is extracted from the fertiliser with a mixture of nitric acid and sulphuric acid.
REAGENTS
3.—3.1 Sulphuric acid (d=1.84 g/ml).
3.2 Nitric acid (d=1.42 g/ml).
APPARATUS
4.—4.1 A Kjeldahl flask, with a capacity of at least 500 ml, or a 250 ml round-bottomed flask with a glass tube forming a reflux condenser.
PREPARATION OF THE SAMPLE
5. See Method 1.
PROCEDURE
Extraction
6.—6.1 Weigh to the nearest 0.001 g, 2.5 g of the prepared sample and place it in a dry Kjeldahl flask (4.1). Add 15 ml water and stir so as to suspend the substance. Add 20 ml nitric acid (3.2) and carefully add 30 ml sulphuric acid (3.1) (see Note). When the initial violent reaction has ceased, slowly bring the contents of the flask to boiling and boil for 30 minutes. Allow to cool and then carefully add with mixing about 150 ml water. Boil for 15 minutes. Cool completely and transfer the liquid quantitively to a 500 ml graduated flask. Make up to volume, mix and filter through a dry fluted filter, discarding the first portion of the filtrate.
Determination
6.2 Determine the phosphorus according to Method 6a or Method 6b on an aliquot part of the clear filtrate.
Note: If the sample contains cellulosic matter, the following procedure is suggested to avoid excessive frothing during digestion:
weigh to the nearest 0.001 g, 2.5 g of the prepared sample and place it in a dry Kjeldahl flask. Add 30 ml sulphuric acid (3.1) and carefully boil until most of the organic matter has been destroyed. Allow to cool, add 15 ml water and 20 ml nitric acid (3.2); bring to the boil and continue boiling for 30 minutes. Continue as described in 6.1 from “Allow to cool and then…”.
5b.EXTRACTION OF PHOSPHORUS BY 2 % CITRIC ACID
SCOPE AND FIELD OF APPLICATION
1. This method is applicable to fertilisers in Groups 2(b), 3(b) and 4(b) of Section A of the Table in Schedule 1 of the Fertilisers Regulations 1990(38) in respect of which an indication of the phosphorus soluble in 2% citric acid is required.
PRINCIPLE
2. The phosphorus is extracted from the fertiliser with a 2% citric acid solution (20 g per litre) in given conditions.
REAGENT
3.—3.1 2% citric acid solution (20 g per litre), prepared from citric acid monohydrate.
APPARATUS
4.—4.1 Rotary shaker: 35-40 turns per minute.
PREPARATION OF THE SAMPLE
5. The analysis is carried out on the product as received after carefully mixing the original sample to ensure it is homogeneous.
See Method 1.
PROCEDURE
Extraction
6.—6.1 Weigh to the nearest 0.001 g, 5 g of the prepared sample, and place it in a dry flask with a sufficiently wide neck, with a capacity of 600 ml, allowing the liquid to be shaken thoroughly. Add 500 ml of the citric acid solution (3.1) at 20 ± 1°C. When adding the first few ml of the reagent, shake vigorously by hand to stop the formation of lumps and to prevent the substance sticking to the sides of the flask. Close the flask with a rubber stopper and shake it in the rotary shaker (4.1) for exactly 30 minutes at a temperature of 20 ± 2°C. Filter immediately through a dry fluted filter into a dry glass receiver and discard the first 20 ml of the filtrate. Continue the filtering until a sufficient quantity of filtrate is obtained to carry out the phosphorous determination.
Determination
6.2 Determine the phosphorus according to Method 6a or Method 6b on an aliquot part of the clear filtrate.
6.DETERMINATION OF EXTRACTED PHOSPHORUS—SPECTROPHOTOMETRIC METHOD
SCOPE AND FIELD OF APPLICATION
1. This method is for the determination of the phosphorus extracted in Methods 5a and 5b.
PRINCIPLE
2. An acidic solution of the extracted phosphorus is treated with molybdovanadate reagent and the absorbance of the yellow solution is measured at 430 nm.
REAGENTS
3.—3.1 Nitric acid (d = 1.42 g/ml).
3.2 Molybdovanadate reagent: dissolve seperately 20 g ammonium molybdate and 0.47 g ammonium vanadate in water, mix, acidify with 140 ml nitric acid (3.1) and dilute to 1 litre with water.
3.3 Phosphorus standard solution: dissolve 4.387 g of potassium dihydrogen phosphate, previously dried at 105°C for 1 hour, in water and dilute to 1 litre.
1 ml of this solution contains 1 mg phosphorus (P) or 2.29 mg phosphorus pentoxide (P2O5).
3.4 Sodium hydroxide, approximately 5 N solution.
APPARATUS
4. Spectrophotometer with 10 mm cells.
PROCEDURE
Determination
5.1.1 For Total Phosphorus
5.1.1Dilute, if necessary, the prepared extract to obtain a phosphorus concentration of about 20 μg/ml. Transfer 10 ml of this solution to a glass stoppered test tube, add 10 ml freshly prepared molybdovanadate reagent (3.2) and mix. Allow to stand for 10 minutes at 20°C and then measure the absorbance of the solution at 430 nm against a freshly prepared reference solution made by adding 10 ml molybdovanadate reagent (3.2) to 10 ml water.
5.1.2 For Water Soluble Phosphorus and Citric Acid Soluble Phosphorus
5.1.2Dilute, if necessary, the prepared extract to obtain a phosphorus concentration of about 80 μg/ml. Transfer 25 ml of this solution to a 100 ml conical flask, add 5 ml nitric acid (3.1) and boil gently for 30 minutes. Cool the solution and neutralise with sodium hydroxide solution (3.4). Cool the solution to 20°C, transfer quantitatively to a 100 ml graduated flask and make up to the mark with water. Transfer 10 ml of this solution to a glass stoppered test tube, add 10 ml freshly prepared molybdovanadate reagent (3.2) and mix. Proceed as described in 5.1.1 from “Allow to stand . . . ”.
Calibration
5.2 From the standard solution (3.3) prepare a series of solutions containing respectively 5, 10, 20, 30 and 40μg/ml of phosphorus (P).
Transfer 10 ml of each solution into a glass-stoppered test tubes, add 10 ml molybdovanadate reagent (3.2), mix and proceed as described in 5.1.1 from “Allow to stand . . .”.
Construct a graph relating the absorbance to the amount of phosphorus present. The calibration curve should be prepared at the same time as the determination is carried out.
EXPRESSION OF RESULTS
6. Determine the amount of phosphorus in the sample by reference to the calibration curve. Express the result in terms of percentage phosphorus (P) or percentage phosphorus pentoxide (P2O5) of the sample:
mg phosphorus (P) × 2.29 = mg phosphorus pentoxide (P205).
7a.DETERMINATION OF POTASSIUM-GRAVIMETRIC METHOD
SCOPE AND FIELD OF APPLICATION
1. This method is applicable to fertilisers in Groups 3(b), 3(c), 3(d) and 4(c) of Section A and Group 5 of Section B of the Table in Schedule 1 of the Fertilisers Regulations 1990(39) in respect of which an indication of total potassium is required.
PRINCIPLE
2. The sample is ashed and dissolved in dilute hydrochloric acid or, if it contains no organic substances, it is dissolved directly in dilute hydrochloric acid. After the removal of interfering substances the potassium is precipitated in a slightly alkaline medium in the form of potassium tetraphenylborate (KTPB).
REAGENTS
3.—3.1 Formaldehyde, 25-35% solution, filtered if necessary before use.
3.2 Potassium chloride.
3.3 Sodium hydroxide, 10 N solution. Care should be taken to ensure that the sodium hydroxide is free from potassium.
3.4 Indicator solution: dissolve 0.5 g phenolphthalein in 100 ml 90% ethanol.
3.5 EDTA solution: 4 g of the dihydrated disodium salt of ethylenediaminetetra-acetic acid (EDTA) per 100 ml. Store this reagent in a plastic container.
3.6 STPB solution: dissolve 32.5 g sodium tetraphenylborate in 480 ml of water, add 2 ml of sodium hydroxide solution (3.3) and 20 ml of a magnesium chloride solution (100 g of MgCl2.6H2O per litre). Stir for fifteen minutes and filter through a fine, ashless filter. Store this reagent in a plastic container.
3.7 Liquid for washing: dilute 20 ml of the STPB solution (3.6) to 1 litre with water.
3.8 Hydrochloricacid(d=1.18g/ml).
APPARATUS
4.—4.1 Filter crucibles with a porosity of 5 to 20 microns.
4.2 Oven regulated at 120°C±1O°C.
PREPARATION OF SAMPLE
5. See Method 1.
PROCEDURE
Preparation of the solution for analysis
6.1.1 Fertilisers containing little or no organic matter
6.1.1Weigh to the nearest 0.001 g, 2.5 g of the prepared sample and transfer to a 400 ml beaker. Add 50 ml water and 5 ml hydrochloric acid (3.8) and evaporate to dryness on a steam bath. Add 5 ml hydrochloric acid (3.8) and 50 ml water. Bring the contents to the boiling point, breaking down any crystals or lumps with a glass rod. Dilute the solution with water to about 100 ml and boil gently for a few minutes. Allow to cool, transfer to a 250 ml graduated flask, dilute to the mark with water and mix; filter through a dry paper.
6.1.2 Fertilisers containing organic matter
6.1.2Weigh to the nearest 0.01 g, 10 g of the prepared sample into a suitable crucible and place in a cold muffle furnace. Gradually raise the temperature to about 475°C (not to exceed 500°C). Maintain at this temperature for at least 16 hours and then open the furnace and allow the crucible to cool. Grind the residue to eliminate any lumps, add 50 ml water and 10 ml hydrochloric acid (3.8) and evaporate to dryness on a steam bath. Proceed as in 6.1.1, commencing “Add 5 ml hydrochloric acid (3.8) and 50 ml water.”.
Deterimination
6.2 6.2.1 Transfer by pipette an aliquot part of the filtrate (6.1.1 or 6.1.2), containing 25-50 mg of potassium (30-60 mg K2O) into a 250 ml beaker; make up to 50 ml with water.
6.2.2To remove interferences, add 10 ml of the EDTA solution (3.5), several drops of the phenolphthalein solution (3.4) and stir in, drop by drop, sodium hydroxide solution (3.3) until it turns red, then finally add a few more drops of sodium hydroxide to ensure an excess (usually I ml of sodium hydroxide is sufficient to neutralise the sample and ensure an excess).
6.2.3To eliminate most of the ammonia boil gently for 15 minutes. Add water to make the volume up to 60 ml. Bring the solution to the boil, remove the beaker from the heat and add 10 ml formaldehyde (3.1). Add several drops of phenolphthalein solution (3.4), and if necessary, more sodium hydroxide solution until a distinct red colour appears. Cover the beaker with a watch glass and place it on a steam bath for fifteen minutes.
Weighing the crucible
6.3 Dry the filter crucible (4.1) to constant weight in the oven at 120°C (4.2) (about 15 minutes).
Allow the crucible to cool in a desiccator and then weigh it.
Precipitation
6.4 Remove the beaker from the steam bath and stir in drop by drop 10 ml of the STPB solution (3.6). This addition should take about 2 minutes; allow to stand for at least 10 minutes before filtering.
Fitlering and washing
6.5 Filter under vacuum into the weighed crucible, rinse the beaker with the liquid for washing (3.7), wash the precipitate three times with the liquid for washing (60 ml in all of the liquid for washing) and twice with 5 to 10 ml of water.
Drying and weighing
6.6 Wipe the outside of the crucible with a filter paper and place in the oven (4.2) for one and a half hours at a temperature of 120°C. Allow the crucible to cool in a desiccator to ambient temperature and weigh rapidly.
Blank test
6.7 Make a blank test under the same conditions (omitting only the sample) and allow for this in the calculation of the final result.
Control test
6.8 Carry out the determination on an aliquot part of an aqueous solution of potassium chloride, containing at the most 40 mg of K2O.
EXPRESSION OF RESULTS
7. Calculate the percentage potassium content of the sample as K2O, taking into account the weight of the test sample, the volume of the aliquot part taken for the determination and the value of the blank determination. (Conversion factor, KTPB to K2O = 0.1314.)
7b.DETERMINATION OF POTASSIUM-FLAME PHOTOMETRIC METHOD
SCOPE AND FIELD OF APPLICATION
1. This method is applicable to fertilisers in Groups 3(b), 3(c), 3(d) and 4(c) of Section A and Group 5 of Section B of the Table in Schedule 1 of the Fertilisers Regulations 1990(40)in respect of which an indication of total potassium is required.
PRINCIPLE
2. The sample is ashed and dissolved in dilute hydrochloric acid or, if it contains no organic substances, it is dissolved directly in dilute hydrochloric acid. The solution is diluted and the potassium content of the extract is determined by flame photometry.
REAGENTS
3.—3.1 Ammonia solution (30%V/V): dilute 30 ml concentrated ammonia solution (d=0.88g/ml) to 100 ml.
3.2 Ammonium oxalate solution: saturated aqueous solution.
3.3 Hydrochloric acid(d=1.18g/ml).
3.4 Potassium dihydrogen phosphate: dried for one hour at 105°C.
3.5 Potassium solution (stock): dissolve 3.4807 g potassium dihydrogen phosphate (3.4) in water and dilute to 1 litre.
3.6 Potassium solution (dilute): dilute 50 ml stock solution (3.5) to 1 litre with water. 1 ml contains 50 potassium (K).
APPARATUS
4.—4.1 Flame photometer.
PREPARATION OF SAMPLE
5. See Method 1.
PROCEDURE
Preparation of the solution for analysis
6.1.1 Fertilisers containing little or no organic matter
6.1.1Weigh to the nearest 0.001 g, 2.5 g of the prepared sample and transfer to a 400 ml beaker. Add 50 ml water and 5 ml hydrochloric acid (3.3) and evaporate to dryness on a steam bath. Add to the residue 125 ml water and 50 ml ammonium oxalate solution (3.2) and boil for 30 minutes. If necessary, a small quantity of potassium-free antifoaming agent may be added. Cool the mixture, add a slight excess of ammonia solution (3.1) and allow to cool. Transfer to a 250 ml graduated flask, dilute to the mark with water, mix and filter through a dry paper.
6.1.2 Fertilisers containing organic matter
6.1.2Weigh to the nearest 0.01 g, 10 g of the prepared sample into a suitable crucible and place in a cold muffle furnace. Gradually raise the temperature to about 475°C (not to exceed 500°C). Maintain at this temperature for at least 16 hours and then open the furnace and allow the crucible to cool. Grind the residue to eliminate any lumps, add 50 ml water and 10 ml hydrochloric acid (3.3) and evaporate to dryness on a steam bath. Add to the residue 125 ml water and 50 ml ammonium oxalate solution (3.2) and boil for 30 minutes. Cool the mixture, add a slight excess of ammonia solution (3.1) and allow to cool. Transfer to a 500 ml graduated flask, dilute to the mark with water, mix and filter through a dry paper.
Blank solution
6.2 Prepare a blank solution from which only the sample has been omitted and allow for this in the calculation of the final results.
Determination
6.3.1 Preparation of sample and blank test solutions
6.3.1Dilute sample solutions (6.1.1 or 6.1.2) and the blank solution (6.2) to a concentration within the optimal measuring range of the flame photometer.
6.3.2 Preparation of the calibration solutions
6.3.2By diluting the standard solution (3.6), prepare at least five standard solutions of increasing concentration corresponding to the optimal measuring range of the flame photometer.
Measurement
6.4 Set the flame photometer to measure the potassium emission according to the manufacturer’s instructions. Spray successively, in triplicate, the standard solutions (6.3.2), the sample solution and the blank solution (6.3. 1), washing the instrument through with distilled water between each spraying. Plot the calibration curve using the median emissions as the ordinates and the corresponding concentrations of potassium in μ/ml as the abscissae. Determine the concentration of potassium in the final sample solution by reference to the calibration curve.
The concentration of potassium in the final solution may be confirmed as follows:
prepare two further dilutions of the standard potassium solution to contain respectively 1 mg/litre more and I mg/litre less than the estimated potassium content of the diluted solution of the sample. Successively spray the low standard solution, the diluted solution of the sample and the high standard solution. Take the median result of each of the three readings and calculate the potassium content of the sample solution.
EXPRESSION OF RESULTS
7. Calculate the percentage potassium content of the sample as K taking into account the weight of the test sample and the dilutions carried out in the course of the analysis. (Conversion factor K to K2O=1.204.)
8.DETERMINATION OF TOTAL MAGNESIUM
EXTRACTION OF TOTAL MAGNESIUM
SCOPE AND FIELD OF APPLICATION
1.—1.1 This method is applicable to all fertilisers.
PRINCIPLE
2.—2.1 Solubilisation by boiling in dilute hydrochloric acid.
REAGENTS
3.—3.1 Diluted hydrochloric acid:
one volume of hydrochloric acid (d=1.18) plus one of water.
APPARATUS
4.—4.1 Electric hot plate with adjustable temperature.
PREPARATION OF THE SAMPLE
5.—5.1 See method 1.
PROCEDURE
Test sample
6.—6.1 Magnesium is extracted from a test sample of five grams weighed to within one milligram.
Preparation of the solution
6.2 Add approximately 400 millilitres of water and, taking care when the sample contains a significant quantity of carbonates, 50 millilitres of dilute hydrochloric acid (4. 1) a small amount at a time. Bring to the boil and maintain for 30 minutes. Allow to cool, stirring occasionally. Decant quantitatively into a 500 millilitre graduated flask. Make up to volume with water and mix. Pass through a dry filter into a dry container, discarding the initial portion. The extract must be completely transparent. Stopper if the filtrate is not used immediately.
DETERMINATION OF MAGNESIUM BY ATOMIC ABSORPTION SPECTRO-PHOTOMETRY
SCOPE AND FIELD OF APPLICATION
1. This method applies to all fertiliser extracts obtained by method 8.1.
PRINCIPLE
2.—2.1 Determination of magnesium by atomic absorption spectrophotometry after appropriate dilution of the extract.
REAGENTS
3.—3.1 Hydrochloric acid, I M solution.
3.2 Hydrochloric acid, 0.5 M solution.
3.3 Standard solution of magnesium, 1.00 mg/ml.
3.3.1Dissolve 1.013 grams of magnesium sulphate (MgSO4.7H2O) in the 0.5 M hydro-chloric acid solution (3.2).
3.3.2Weigh out 1.658 grams of magnesium oxide (MgO), previously calcined to remove all traces of carbonation. Place in a beaker with 100 ml of water and 120 ml of I M hydrochloric acid (3. 1). When it has dissolved, decant quantitatively into a 1,000 ml graduated flask. Make up the volume by adding and mix
or
3.3.3 Commercial standard solution
3.3.3The laboratory is responsible for testing such solutions.
Strontium chloride solution
3.4 Dissolve 75 grams of strontium chloride (SrCl2. 6H2O) in a hydrochloric acid solution (3.2) and make up to 500 ml with the same acid solution.
APPARATUS
4.—4.1 Spectrophotometer fitted for atomic absorption, with a magnesium lamp, set at 285.2 nm.
4.2 Air-acetylene flame.
PREPARATION OF THE SAMPLE
5.—5.1 See Method 8.1.
PROCEDURE
6.—6.1 If the fertiliser has a declared magnesium (Mg) content of more than 6% (ie 10% as MgO), take 25 ml (V1) of the extraction solution (5). Transfer into a 100 ml graduated flask, and make up to volume with water and mix. The dilution factor is D1=1OO/V1.
6.2 Using a pipette, take 10 millilitres of the extraction solution (5) or the solution (6.1). Transfer into a 200 ml graduated flask. Make up to volume with the 0.5 M hydrochloric acid solution (3.2) and mix. The dilution factor is 200/10.
6.3 Dilute this solution (6.2) with the 0.5 M hydrochloric acid solution (3.2) so as to obtain a concentration in the optimum working field of the spectrophotometer (4.1). V2 is the volume of the sample in 100 ml. The dilution factor is D2= 100/V 2.
The final solution should contain 10% v/v of the strontium chloride solution (3.4).
Preparation of the blank solution
6.4 Prepare a blank solution by repeating the whole procedure from the extraction (method 8.1), omitting only the test sample of fertiliser.
Preparation of calibration solutions
6.5 By diluting the standard solution (3.3) with the 0.5 M hydrochloric acid, prepare at least five calibration solutions of increasing concentration within the optimum measuring range of the apparatus (4.1).
These solutions should contain 10% v/v of the strontium chloride solution (3.4).
Measurement
6.6 Set up the spectrophotometer (4.1) at a wavelength of 285.2 nm.
Spray, successively, the calibration solutions (6.5), the sample solution (6.3) and the blank solution (6.4), washing the instrument through with the solution to be measured next. Repeat this operation three times. Plot the calibration curve using the mean absorbances of each of the calibrations (6.5) as the ordinates and the corresponding concentration of magnesium in μ/ml as the abscissae. Determine the concentration of magnesium in the sample (6.3), Xs, and blank (6.4), Xb, by reference to the calibration curve.
EXPRESSION OF RESULTS
7. Calculate the amount of magnesium (Mg) or magnesium oxide (MgO) in the sample by reference to the calibration solutions and taking into consideration the blank.
The percentage of magnesium (Mg)in the fertiliser is equal to:

where:
Xs= the concentration of the solution to be analysed recorded on the calibration curve, in μ/ml.
Xb= the concentration of the blank solution as recorded on the calibration curve, in μ/ml.
D1= the dilution factor when the solution is diluted (6. 1).
It is equal to four if 25 ml are taken.
It is equal to one when the solution is not diluted.
D2= the dilution factor in 6.3.
M= the mass of the test sample at the time of extraction.
MgO(%)=Mg(%)/0.6.
9a.DETERMINATION OF BORON-TITRIMETRIC METHOD
SCOPE AND FIELD OF APPLICATION
1. This method is applicable to all fertilisers where the levels of boron are greater than 1,000 mg/kg.
PRINCIPLE
2. The sample is dissolved in acid, the solution treated with lead nitrate in order to remove phosphate and the borate in the filtrate is titrated potentiometrically in the presence of mannitol.
REAGENTS
3.—3.1 Calcium oxide.
3.2 Mannitol.
3.3 Sodium carbonate.
3.4 Hydrochloric acid solution 50% (V/V): dilute 50 ml concentrated hydrochloric acid (d = 1.181 g/ml) with water to 100 ml.
3.5 Hydrochloric acid, 0.5 N solution.
3.6 Lead nitrate solution, 10 g per 100 ml.
3.7 Sodium hydroxide, 0.5 N solution.
3.8 Sodium hydroxide, 0.05 N solution, carbonate free.
3.9 Methyl red indicator solution: dissolve 0. 1 g of methyl red in 50 ml 95% ethanol, make up to 100 ml with water and filter if necessary.
3.10 Phenolphthalein indicator solution: dissolve 0.25 g phenolphthalein in 150 ml 95% alcohol and dilute with water to 250 ml.
APPARATUS
4.—4.1 pH meter.
PREPARATION OF SAMPLE
5. See Method 1.
PROCEDURE
Preparation of the solution for analysis
6.1.1 In the absence of organic matter
6.1.1Weigh to the nearest 0.001 g, 2 g of the prepared sample if the boron content is 0.5% or less, or 1 g if the boron content is from 0.5 — 1.0%, and place in a 400 ml beaker. Add I 00 ml water, a few drops of phenolphthalein indicator solution (3.10) and sufficient sodium carbonate (3.3) to make the solution slightly alkaline. Boil gently and keep the boiling solution alkaline, adding more sodium carbonate (3.3) as necessary until all the ammonia which may be present has been evolved. Cool the solution and add 12 ml hydrochloric acid solution (3.4).
6.1.2 In the presence of organic matter
6.1.2Weigh to the nearest 0.001 g, 2 g of the prepared sample if the boron content is 0.5% or less, or 1 g if the boron content is from 0.5 — 1.0%, and place it in a silica dish. Add 0.2 g calcium oxide (3. 1) for each 1 g of sample, moisten with water, mix thoroughly, evaporate the mixture to dryness and transfer the crucible to a cold muffle furnace. Raise the temperature slowly to 450±10°C and then ignite for about 3 hours. Remove the crucible from the furnace, cool and moisten the ash with 10 ml of hydrochloric acid solution (3.4). Warm the solution on a steam bath for 15 minutes, covering the dish with a watch glass. Transfer the contents of the dish quantatively into a 400 ml beaker, add a few drops of phenolphthalein indicator solution (3.10) and dilute to about 120 ml with water.
Determination
6.2 To the prepared solution (6. 1.1 or 6.1.2), add 20 ml lead nitrate solution (3.6) for each 12% P2O, in the sample if 2 g of the sample has been used; add I 0 ml lead nitrate solution for each 12% P2O, in the sample if 1 g of the sample has been used. Heat to boiling, remove from the source of heat and make slightly alkaline by addition of sodium carbonate (3.3). Warm the solution on a steam bath for five minutes, cool and transfer the solution quantitatively into a 200 ml graduated flask. Make up to the mark with water, mix and filter through a 24 cm filter paper(1), rejecting the first 10-21 ml of the filtrate.
Transfer 100 ml of the filtrate into a 250 ml beaker, add a few drops of methyl red indicator (3.9) and acidify the solution with 0.5 N hydrochloric acid solution (3.5). Heat almost to boiling, stir vigorously to remove carbon dioxide, keeping the solution acidic, by adding if necessary more 0.5 N hydrochloric acid solution (3.5). Neutralise the solution with 0.5 N sodium hydroxide solution (3.7), and then make just acid by addition of 0.5 N hydrochloric acid solution (3.5). Cover the beaker with a watch glass and boil the solution gently for 5 minutes in order to expel any remaining carbon dioxide.
Cool the solution rapidly and using the pH meter (4.1), adjust the pH of the solution to 6.3 by the addition of 0.05 N sodium hydroxide solution (3.8). Add 10 g mannitol (3.2) and titrate the solution with 0.05 N sodium hydroxide solution to a pH of 6.3. Continue to add further 10 g portions of mannitol (3.2) and to re-adjust the pH to 6.3 until after the final addition of mannitol the pH remains constant at 6.3. The total amount of 0.05 N sodium hydroxide solution used after the first addition of mannitol corresponds to the amount of boron present in the sample solution. Allow a standard value of 0.1 ml 0.05 N sodium hydroxide solution as ’blank’ value.
EXPRESSION OF RESULT
7. The percentage boron content of the sample is given by the formula

where:
T = ml of 0.05 N sodium hydroxide solution used after the addition of the mannitol
M = weight of the sample in grams.
9b.DETERMINATION OF BORON-SPECTROPHOTOMETRIC METHOD
SCOPE AND FIELD OF APPLICATION
1. This method is applicable to all fertilisers for levels of boron up to 1,000 mg/kg.
PRINCIPLE
2. The sample is ashed in the presence of calcium oxide and the residue is dissolved in hydrochloric acid. The resulting solution is treated with carmine to form a coloured complex with boron, the absorption of which is measured at 625 nm.
REAGENTS
3.—3.1 Calcium oxide.
3.2 Sulphuric acid (d= 1.84 g/ml).
3.3 Carminic acid solution: dissolve 0.025 g carminic acid in sulphuric acid (3.2) and dilute to 100 ml with sulphuric acid (3.2).
3.4 Hydrochloric acid solution 20% (V/V): dilute 20 ml hydrochloric acid (d = 1. 1 8 g/ml) with water to 100 ml.
3.5.1Boron solution (stock):
weigh to the nearest 0.001 g, 1.905 g boric acid, dissolve in water and dilute to 1 litre with water.
1 ml of this solution = 0.333 mg boron.
3.5.2Boron solution (working standard):
dilute 10 ml of boric acid stock solution (3.5. 1) with water to 100 ml.
Transfer 5, 10, 15, 20 and 25 ml respectively into separate 100 ml graduated flasks and dilute to the marks with water. These solutions contain 5, 10, 15, 20 and 25ug of boron per 3 ml of solution.
3.6 Hydrazine hydrate (approximately 60% W/W solution).
WARNING: Hydrazine hydrate is toxic and corrosive, causing burns; avoid contact with eyes and skin.
APPARATUS
4.—4.1 Spectrophotometer with 10 mm cells.
PREPARATION OF THE SAMPLE
5. See Method 1.
PROCEDURE
Preparation of the solution for analysis
6.—6.1 Weigh to the nearest 0.001 g, 5 g of the prepared sample and place it in a silica dish. Add 1 g calcium oxide (3.1), moisten with water, mix thoroughly, evaporate the mixture to dryness and then transfer the crucible to a cold muffle furnace. Raise the temperature slowly to 450±10°C and ignite for about 3 hours. Remove the crucible from the furnace, cool and add hydrochloric acid solution (3.4) until the resulting mixture is acid, then add 5 ml hydrochloric acid solution (3.4) in excess. Heat the mixture at 70°C for 15 minutes, cool and filter through a filter paper(2) into a suitable graduated flask washing both the dish and the filter with water. Make up to the mark with water and mix. Dilute an aliquot of this solution so that 3 ml contains between 5 and 25 μ of boron.
Blank test
6.2 Carry out a blank test omitting only the sample.
Determination
6.3 Transfer 3 ml of the prepared solution (6.1) to a small conical flask, add cautiously 15 ml sulphuric acid (3.2), swirl the flask and add 10 ml carminic acid solution (3.3). Cool the flask rapidly to room temperature, mix well and allow to stand for 2 hours. Measure the absorbance in the spectrophotometer (4.1) at 625 nm with water as reference. Determine the quantity of boron in the solution by reference to the calibration curve (6.4).
Calibration curve
6.4 Transfer 3 ml of each working standard solution (3.5.2) into a series of small conical flasks and proceed as described in 6.3 commencing at “ . . add cautiously 15 ml sulphuric acid (3.2). . .”. Plot a calibration curve of the absorbance of the solution against the corresponding quantities of boron, μg.
EXPRESSION OF RESULTS
7. The boron content in mg/kg is given by the formula:

where:
A = weight of boron in the 3 ml aliquot taken for colour development after allowing for the blank reading (μ)
V = volume of prepared solution before dilution
F = factor allowing for dilution under 6.1
M = weight of the sample in grams.
Note: The colour of the boron-carmine complex is affected by nitrate nitrogen. When this form of nitrogen is known to be present in the sample, the “Determination” procedure (6.3) should be modified by the addition of 0. 5 ml hydrazine hydrate (3.6) before the addition of the sulphuric acid which should be carried out under conditions of extreme caution because of the (3.2) violent nature of the reaction. (The use of a burette for the addition of the sulphuric acid is recommended.) The hydrazine hydrate (3.6) has no influence on the absorption of the boron-carmine complex and therefore is not added to the standard solutions in the preparation of the calibration curve. However, in order to equalise the volumes of the solutions of both samples and standards, add 0.5 ml water to the latter.
10.DETERMINATION OF COBALT
SCOPE AND FIELD OF APPLICATION
1. This method is applicable to all fertilisers.
PRINCIPLE
2. The sample is dissolved in hydrochloric acid (after ashing if necessary) and the solution is treated with citric acid in order to prevent precipitation of iron and phosphate. Cobalt is extracted as its 2-nitroso-1-naphthol complex into toluene. The cobalt content is measured at 367 nm, by reference to a calibration curve.
REAGENTS
3.—3.1 Sodium sulphate, anhydrous.
3.2 Toluene.
3.3 Hydrochloric acid, 2 N solution.
3.4 Hydrochloric acid solution, 50— (V/V): dilute 50 ml concentrated hydrochloric acid solution (d = 1.18 g/ml) to 100 ml with water.
3.5 Hydrogen peroxide solution, 3% (10 volume).
3.6 Nitric acid solution, 30% (V/V): dilute 30 ml nitric acid (d = 1.42 g/ml) with water to 1OOml.
3.7 2-nitroso-1-naphthol solution:
dissolve 1 g of 2-nitroso-l-naphthol in 100 ml glacial acetic acid and add 1 g activated carbon.
Shake the solution before use and filter off the required amount.
3.8 Sodium citrate solution: 40 g per 100 ml.
3.9 Sodium hydroxide, 2 N solution.
3.10 Cobalt solution (stock):
weigh to the nearest 0.001 g, 0.670 g ammonium cobaltous sulphate, [(NH1)2CO(SO4)2.6H2O]
dissolve in water and make up to 100 ml with water. 1 ml of this solution contains 1,000μ cobalt.
3.11 Cobalt solution (working standard):
dilute the stock colbalt solution (3.10) as required so that 1 ml contains 1μ cobalt. Prepare the solution freshly before use.
APPARATUS
4.—4.1 Spectrophotometer with 10 mm cells.
PREPARATION OF THE SAMPLE
5. See Method 1.
PROCEDURE
Preparation of the solution for analysis
6.1.1 In the absence of organic matter
6.1.1Weigh to the nearest 0.001 g, 5 g of the prepared sample, place in a 100 ml beaker, add 10 ml hydrochloric acid solution (3.4) and evaporate to dryness on a steam bath. Extract the soluble salts with three successive 10 ml portions of boiling 2 N hydrochloric acid solution (3.3), decanting the solution each time through the same filter paper(3) into a 50 ml graduated flask. Wash the filter paper with a little water, cool the solution to room temperature and make up to the mark with water.
6.1.2 In the presence of organic matter
6.1.2Weigh to the nearest 0.001 g, 5 g of the prepared sample into a silica dish and place a silica cover on top. Transfer the dish to a cold mute furnace, raise the temperature to about 475°C (do not exceed 500°C). Maintain at this temperature for at least 16 hours and then open the furnace and allow the crucible to cool. Add 10 ml hydrochloric acid solution (3.4) and evaporate to dryness on a steam bath. Extract the soluble salts with two successive 10 ml portions of boiling 2 N hydrochloric acid solution (3.3), decanting the solution each time through the same filter paper(4) into a 50 ml graduated flask. Add 5 ml hydrochloric acid solution (3.4) and 5 ml nitric acid solution (3.6) to the residue in the dish and evaporate the mixture to dryness on a hot plate at low heat. Add 10 ml boiling hydrochloric acid solution (3.3) to the residue and filter the solution through the same filter paper into the 50 ml graduated flask. Wash the filter paper with water, cool the solution to room temperature and make up to the mark with water.
Determination
6.2 6.2.1 Transfer a suitable aliquot of the solution prepared in 6.1 (containing not more than 15 μ cobalt) to a 100 ml beaker, add 15 ml sodium citrate solution (3.8), dilute to about 50 ml with water and adjust the pH to between 3 and 4 by adding 2 N hydrochloric acid solution (3.3). (A precipitate of ferric hydroxide may form but this can be dissolved by heating the solution.) Cool to room temperature, add 10 ml hydrogen peroxide solution (3.5) and, after 5 minutes, 1 ml 2-nitroso-l-naphthol solution (3.7). Heat the solution to about 90°C and then allow to stand for 30 minutes at room temperature. Transfer the solution to a 125 ml separating funnel, add I 0 ml toluene (3.2), shake vigorously for 2 minutes, allow the phases to separate and discard the lower aqueous phase.
6.2.2To the toluene extract add 20 ml 2 N hydrochloric acid solution (3.3), shake for 1 minute and discard the lower aqueous phase. Add 20 ml 2 N sodium hydroxide solution (3.9), shake for 1 minute and again discard the aqueous phase. Repeat the washing with a further 20 ml of 2 N sodium hydroxide solution (3.9). Finally run off the toluene solution through a little anhydrous sodium sulphate (3.1) into a clean dry stoppered tube. Carry out a blank determination repeating the procedure, but omitting the sample. Measure the absorbance of the magenta coloured solutions at a wave length of 367 nm in the spectrophotometer (4.1) with toluene (3.2) as reference. Determine the quantity of cobalt in the solution by reference to the calibration curve (6.3).
Calibration curve
6.3 Measure amounts of cobalt working standard solution (3.1 1) corresponding to 3, 6, 9, 12 and 15μ cobalt into five separate 100 ml beakers and proceed as described in 6.2 commencing at “. . . add 15 ml sodium citrate solution (3.8). . .”. Plot a calibration graph of the absorbance of the solutions against the corresponding amounts of cobalt (μ).
EXPRESSION OF RESULTS
7. The cobalt content in mg/kg is given by formula:

where:
A = weight of cobalt taken for colour development as read from the calibration graph after allowing for the blank reading (mgr;)
V = volume in ml of sample taken for colour development
M = weight of sample in grams.
11DETERMINATION OF MOLYBDENUM
SCOPE AND FIELD OF APPLICATION
1. This method is applicable to all fertilisers.
PRINCIPLE
2. The sample is dissolved in hydrochloric acid (after ashing if necessary) and the molybdenum complexed with thiocyanate in the presence of stannous chloride. The red coloured complex is extracted into an organic solvent mixture and its absorbance measured at 470 nm.
REAGENTS
3.—3.1 Hydrochloric acid, 50% (V/V): dilute 50 ml concentrated hydrochloric acid solution (d= 1. 18 g/ml) to 100 ml with water.
3.2 Hydrochloric acid, 2 N solution.
3.3 Hydrochloric acid, N solution.
3.4 Nitric acid solution, 30% (V/V): dilute 30 ml nitric acid (d= 1.42 g/ml) with water to 100 ml.
3.5.1Molybdenum solution (working standard):
dissolve 1.84 g ammonium molybdate [(NH4)6Mo7O24.4H2O] in water and dilute with water to 1 litre.
3.5.2Molybdenum solution (working standard):
dilute 1.0 ml stock solution (3.5.1) to 1 litre with water.
(1 ml = 1μ molybdenum).
Prepare this solution immediately prior to use.
3.6 Ammonium ferrous sulphate solution, 4 g per 100 ml.
3.7 Potassium thiocyanate solution, 40 g per 100 ml.
3.8 Sodium sulphate, anhydrous.
3.9 Stannous chloride solution: suspend 40 g stannous chloride dihydrate in 20 ml 6.5 N hydrochloric acid, add water to dissolve and dilute to 100 ml. Filter if turbid.
3.10 Solvent mixture: mix equal volumes of carbon tetrachloride and 3-methylbutan-1-ol.
APPARATUS
4.—4.1 Spectrophotometer with 10 mm cells.
PREPARATION OF THE SAMPLE
5. See Method 1.
PROCEDURE
Preparation of the solution for analysis
6.1.1 In the absence of organic matter
6.1.1Weigh to the nearest 0.001 g, 5 g of the prepared sample, place in a 100 ml beaker, add 10 ml hydrochloric acid solution (3.1) and evaporate to dryness on a steam bath. Extract the soluble salts with three successive 10 ml portions of boiling 2 N hydrochloric acid solution (3.2), decanting the solution each time through the same filter paper(5) into a 50 ml graduated flask. Wash the filter paper with a little water, cool the solution to room temperature and make up to the mark with water.
6.1.2 In the presence of organic matter
6.1.2Weigh to the nearest 0.001 g, 5 g of the prepared sample into a silica dish and place a silica cover on top. Transfer the dish to a cold muffle furnace and gradually raise the temperature to about 475°C (not to exceed 500°C). Maintain at this temperature for at least 16 hours and then open the furnace and allow the crucible to cool. Add 10 ml hydrochloric acid solution (3.1) and evaporate to dryness on a steam bath. Extract the soluble salts with two successive 10 ml portions of boiling 2 N hydrochloric acid solution (3.2), decanting the solution each time through the same filter paper(5) into a 50 ml graduated flask. Add 5 ml hydrochloric acid solution (3.1) and 5 ml nitric acid solution (3.4) to the residue in the dish and evaporate the mixture to dryness on a hot plate at low heat. Add 10 ml boiling hydrochloric acid solution (3.1) to the residue and filter the solution through the same filter paper into the 50 ml graduated flask. Wash the filter paper with water, cool the solution to room temperature and make up to the mark with water.
Determination
6.2 6.2.1 Transfer a suitable aliquot of the solution, prepared as in 6.1, to a 125 ml separating funnel, add I ml ammonium ferrous sulphate solution (3.6) and sufficient N hydrochloric acid (3.3) to bring the volume to 50 ml (see Note), then add 1 ml potassium thiocyanate solution (3.7) and mix. Add 1 ml stannous chloride solution (3.9) and mix again. Add exactly 7 ml solvent mixture (3.10), shake vigorously for two minutes and allow to separate for fifteen minutes. Filter the lower layer through a 7 cm paper into a small stoppered tube. (If the lower layer is not clear of if filtration is difficult, filter through a suitable column packed with anhydrous sodium sulphate (3.8), solid stannous chloride and plugged with cotton wool.)
6.2.2Carry out a blank determination repeating the procedure but omitting the sample. Measure the absorbance of the solutions at a wave length of 470 nm, in the spectrophotometer (4.1) with water as reference. Determine the quantity of molybdenum in the solution by reference to the calibration curve (6.3).
Note: The acidity of final solution must not exceed 1.5 N with respect to hydrochloric acid; with more strongly acid conditions, fading of the colour will occur.
Calibration curve
6.3 Transfer by pipette, 0, 5, 10, 15, 20 and 25 ml standard molybdenum solution (3.5.2) into a series of 125 ml separating funnels. To each funnel add 1 ml ammonium ferrous sulphate solution (3.6) and 25 ml of 2 N hydrochloric acid (3.2); dilute to 50 ml with water where necessary and proceed as described at 6.2.1, commencing at “then add I ml potassium thiocyanate solution (3.7) and mix. . .”. Plot a calibration curve of the absorbance of the solutions against the corresponding amounts of molybdenum (μg).
EXPRESSION OF RESULTS
7. The molybdenum content in mg/kg is given by the formula:
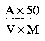
where:
A = weight of molybdenum in the aliquot taken for colour development as read from the calibration curve after allowing for the blank reading (μg)
V = volume in ml of aliquot taken for colour development
M = weight of sample in grams.
12.DETERMINATION OF COPPER
SCOPE AND FIELD OF APPLICATION
1. This method is applicable to all fertilisers.
PRINCIPLE
2. The sample is ashed and dissolved in dilute hydrochloric acid or, if it contains no organic substances, it is dissolved directly in dilute hydrochloric acid. The solution is diluted and the copper content is determined by atomic absorption spectrophotometry.
REAGENTS
3.—3.1 Hydrochloric acid (d = 1. 18 g/ml).
3.2 Hydrochloric acid, 6 N solution.
3.3 Hydrochloric acid, 0.5 N solution.
3.4 Hydrogen peroxide, approximately 100 volume, 30% by weight.
3.5.1Copper solution(6) (stock):
weigh to the nearest 0.001 g, 1 g pure copper, dissolve in 25 ml 6 N hydrochloric acid solution (3.2), add 5 ml hydrogen peroxide (3.4) and dilute to 1 litre with water. 1 ml of this solution = 1,000 μ of copper (Cu).
3.5.2Copper solution (dilute):
dilute 10 ml of stock solution (3.5.1) to 100 ml with water and then dilute the resulting solution, 10 ml to I 00 ml with water. 1 ml of the final dilution = 10μ of copper (Cu).
APPARATUS
4.—4.1 Atomic absorption spectrophotometer with a copper lamp (324.8 nm).
PREPARATION OF SAMPLE
5. See Method 1.
PROCEDURE
Preparation of the solution for analysis
6.1.1 In the absence of organic matter
6.1.1Weigh to the nearest 0.001 g, 5 g of the prepared sample, place it in a 400 ml beaker, add carefully 5 ml hydrochloric acid (3.1) (there may be a vigorous reaction due to carbon dioxide formation). Add more hydrochloric acid, if necessary. When effervescence has stopped, evaporate to dryness on a steam bath, stirring occasionally with a glass rod. Add 15 ml 6 N hydrochloric acid solution (3.2) and 120 ml water. Stir with the glass rod, which should be left in the beaker, and cover the beaker with a watch glass. Boil the solution gently until dissolution appears complete and then filter through a filter paper(7) into a 250 ml graduated flask. Wash the beaker and filter with 5 ml hot 6 N hydrochloric acid solution (3.2) and twice with boiling water. Cool and make up to the mark with water (the hydrochloric acid concentration of this solution should be about 0.5 N).
6.1.2 In the presence of organic matter
6.1.2Weigh to the nearest 0.001 g, 5 g of the prepared sample into a silica or platinum crucible and place the crucible into a cold mute furnace. Close the furnace and gradually raise the temperature to 450-475°C over about 90 minutes. Maintain this temperature for at least 16 hours and then open the furnace and allow the crucible to cool. Moisten the ash with water and transfer it into a 250 ml beaker. Wash the crucible with about 5 ml hydrochloric acid (3. 1) and add the latter slowly and carefully to the beaker (there may be a vigorous reaction due to carbon dioxide formation). If necessary, add more hydrochloric acid (3.1) with stirring, until all effervescence has stopped. Evaporate the solution to dryness, occasionally stirring with a glass rod. Add 15 ml 6 N hydrochloric acid solution (3.2) and 120 ml water. Stir with the glass rod, which should be left in the beaker, and cover with a watch glass. Boil the solution gently until dissolution appears complete and filter through a filter paper(7) into a 250 ml graduated flask. Wash the beaker and filter with 5 ml of hot 6 N hydrochloric acid solution (3.2) and twice with boiling water. Cool and make up to the mark with water. (The hydrochloric acid concentration of this solution should be about 0.5 N.)
Blank solution
6.2 Prepare a blank solution from which only the sample has been omitted and allow for this in the calculation of the final results.
Determination
6.3.1 Preparation of sample and blank test solutions
6.3.1Dilute the sample solutions (6.1.1 or 6.1.2) and the blank test solution (6.2) with 0.5 N hydrochloric acid solution (3.3) to a concentration within the optimal measuring range of the spectrophotometer.
6.3.2 Preparation of the calibration solution
6.3.2By diluting the standard solution (3.5.2) with 0. 5 N hydrochloric acid solution (3.3) prepare at least 5 standard solutions corresponding to the optimal measuring range of the spectrophotometer.
Measurement
6.4 Set up the spectrophotometer (4.1) at a wavelength of 324.8 nm using an oxidising air-acetylene flame. Spray successively, in triplicate, the standard solution (6.3.2), the sample solution and the blank test solution (6.3.1), washing the instrument through with distilled water between each spraying. Plot the calibration curve using the mean absorbances as the ordinates and the corresponding concentrations of copper μ/ml as the abscissae.
Determine the concentration of copper in the final sample and blank solution by reference to the calibration curve.
EXPRESSION OF RESULTS
7. Calculate the copper content of the sample taking into account the weight of the test sample and the dilutions carried out in the course of the analysis. Express the result either as a percentage or as mg/kg.
13.DETERMINATION OF IRON
SCOPE AND FIELD OF APPLICATION
1. This method is applicable to all fertilisers.
PRINCIPLE
2. The sample is ashed and dissolved in dilute hydrochloric acid or, if it contains no organic substances, it is dissolved directly in dilute hydrochloric acid. The solution is diluted and the iron content of the extract is determined by atomic absorption spectrophotometry.
REAGENTS
3.—3.1 Hydrochloric acid (d= 1.18 g/ml).
3.2 Hydrochloric acid, 6 N solution.
3.3 Hydrochloric acid, 0.5 N solution.
3.4 Hydrogen peroxide, approximately 100 volume, 30% by weight.
3.5.1Iron solution(8) (stock):
weigh to the nearest 0.001 g, 1 g pure iron, dissolve in 200 ml 6 N hydrochloric acid solution (3.2), add 16 ml hydrogen peroxide solution (3.4) and dilute to 1 litre with water.
1 ml of this solution = 1,000 μ of iron (Fe).
3.5.2Iron solution (dilute):
dilute 10 ml of stock solution (3.5.1) to 100 ml with water.
1 ml of this solution = 100 μ of iron (Fe).
3.6 Lanthanum chloride solution:
dissolve 12 g lanthanum oxide in 150 ml water, add 100 ml 6 N hydrochloric acid solution (3.2) and dilute to 1 litre with water.
APPARATUS
4.—4.1 Atomic absorption spectrophotometer with an iron lamp (248.3 nm).
PREPARATION OF SAMPLE
5. See Method 1.
PROCEDURE
Preparation of the solution for analysis
6.1.1 In the absence of organic matter
6.1.1Weigh to the nearest 0.001 g, 5 g of the prepared sample, place it in a 400 ml beaker, add carefully 5 ml hydrochloric acid (3. 1) (there may be a vigorous reaction due to carbon dioxide formation). Add more hydrochloric acid, if necessary. When effervescence has stopped, evaporate to dryness on a steam bath, stirring occasionaly with a glass rod. Add 15 ml 6 N hydrochloric acid solution (3.2) and 120 ml water. Stir with the glass rod, which should be left in the beaker, and cover the beaker with a watch glass. Boil the solution gently until dissolution appears complete and then filter through a filter paper(9) into a 250 ml graduated flask. Wash the beaker and filter with 5 ml of hot 6 N hydrochloric acid solution (3.2) and twice with boiling water. Cool and make up to the mark with water. (The hydrochloric acid concentration of this solution should be about 0.5 N.)
6.1.2. In the presence of organic matter
6.1.2.Weigh to the nearest 0.001 g, 5 g of the prepared sample into a silica or platinum crucible and place the crucible in a cold muffle furnace. Close the furnace and gradually raise the temperature to 450-475° over about 90 minutes. Maintain this temperature for at least 16 hours and then open the furnace and allow the crucible to cool. Moisten the ash with water and transfer it into a 250 ml beaker. Wash the crucible with about 5 ml hydrochloric acid (3.1) and add the latter slowly and carefully to the beaker (there may be a vigorous reaction due to carbon dioxide formation). If necessary, add more hydrochloric acid (3.1) with stirring, until all effervescence has stopped.
Evaporate the solution to dryness, occasionally stirring with a glass rod. Add 15 ml 6 N hydrochloric acid solution (3.2) and 120 ml water. Stir with the glass rod, which should be left in the beaker, and cover with a watch glass. Boil the solution gently until dissolution appears complete and filter through a filter paper(10) into a 250 ml graduated flask. Wash the beaker and filter with 5 ml of hot 6 N hydrochloric acid solution (3.2) and twice with boiling water. Cool and make up to the mark with water. (The hydrochloric acid concentration of this solution should be about 0.5 N.)
Blank solution
6.2 Prepare a blank solution from which only the sample has been omitted and allow for this in the calculation of the final results.
Determination
6.3.1 Preparation of sample and blank test solutions
6.3.1Dilute the sample solutions (6.1.1 or 6.1.2) and the blank test solution (6.2) with 0.5 N hydrochloric acid solution (3.3) to a concentration within the optimal measuring range of the spectrophotometer. The final solution must contain 10% (V/V) of the lanthanum chloride solution (3.6).
6.3.2 Preparation of the calibration solutions
6.3.2By diluting the standard solution (3.5.2) with 0.5 N hydrochloric acid solution (3.3) prepare at least 5 standard solutions of increasing concentration corresponding to the optimal measuring range of the spectrophotometer. The final solutions must contain 10% (V/V) of the lanthanum chloride solution (3.6).
Measurement
6.4 Set up the spectrophotometer (4.1), at a wave length of 248.3 nm using an oxidising air-acetylene flame. Spray successively, in triplicate, the standard solutions (6.3.2), the sample solution, and the blank test solution (6.3.1), washing the instrument through with distilled water between each spraying. Plot the calibration curve using the mean absorbances as the ordinates and the corresponding concentrations of iron in υ/ml as the abscissae. Determine the concentration of iron in the final sample and blank solutions by reference to the calibration curve.
EXPRESSION OF RESULTS
7. Calculate the iron content of the sample taking into account the weight of the test sample and the dilutions carried out in the course of the analysis. Express the result either as a percentage or as mg/kg.
14.DETERMINATION OF MANGANESE
SCOPE AND FIELD OF APPLICATION
1. This method is applicable to all fertilisers.
PRINCIPLE
2. The sample is ashed and dissolved in dilute hydrochloric acid or, if it contains no organic substances, it is dissolved directly in dilute hydrochloric acid. The solution is diluted and the manganese content of the extract is determined by atomic absorption spectrophotometry.
REAGENTS
3.—3.1 Hydrochioricacid(d=1.18g/ml).
3.2 Hydrochloric acid, 6 N solution.
3.3 Hydrochloric acid, 0.5 N solution.
3.4.1Manganese solution(11) (stock):
weigh to the nearest 0.001 g, 1g pure manganese, dissolve in 25 ml 6 N hydrochloric acid solution (3.2) and dilute to 1 litre with water. 1 ml of this solution= 1,000 μ of manganese (Mn).
3.4.2Manganese solution (dilute):
dilute 10 ml of stock solution (3.4.1) to 1 litre with water. 1 ml of this solution=10 μ of manganese.
3.5 Lanthanum chloride solution:
dissolve 12 g lanthanum oxide in 150 ml water, add 100 ml 6 N hydrochloric acid solution (3.2) and dilute to 1 litre with water.
APPARATUS
4.—4.1 Atomic absorption spectrophotometer with a manganese lamp (279.5 nm).
PREPARATION OF SAMPLE
5. See Method 1.
PROCEDURE
Preparation of the solution for analysis
6.1.1 In the absence of organic matter
6.1.1Weigh to the nearest 0.001 g, 5 g of the prepared sample, place it in a 400 ml beaker, add carefully 5 ml hydrochloric acid (3.1) (there may be a vigorous reaction due to carbon dioxide formation). Add more hydrochloric acid, if necessary. When effervescence has stopped, evaporate to dryness on a steam bath, stirring occasionally with a glass rod. Add 15 ml 6 N hydrochloric acid solution (3.2) and 120 ml water. Stir with the glass rod, which should be left in the beaker, and cover the beaker with a watch glass. Boil the solution gently until dissolution appears complete and then filter through a filter paper(12) into a 250 ml graduated flask. Wash the beaker and filter with 5 ml of hot 6 N hydrochloric acid solution (3.2) and twice with boiling water. Cool and make up to the mark with water. (The hydrochloric acid concentration of this solution should be about 0.5 N.)
6.1.2 In the presence of organic matter
6.1.2Weigh to the nearest 0.001 g, 5 g of the prepared sample into a silica or platinum crucible and place the crucible into a cold muffle furnace. Close the furnace and gradually raise the temperature to 450-475°C over about 90 minutes. Maintain this temperature for at least 16 hours and then open the furnace and allow the crucible to cool. Moisten the ash with water and transfer it into a 250 ml beaker. Wash the crucible with about 5 ml hydrochloric acid (3. 1) and add the latter slowly and carefully to the beaker (there may be a vigorous reaction due to carbon dioxide formation). If necessary, add more hydrochloric acid (3. 1) with stirring, until all effervescence has stopped. Evaporate the solution to dryness, occasionally stirring with a glass rod. Add 15 ml of 6 N hydrochloric acid solution (3.2) and 120 ml water. Stir with the glass rod, which should be left in the beaker, and cover with a watch glass. Boil the solution gently until dissolution appears complete and filter through a filter paper(12) into a 250 ml graduated flask. Wash the beaker and filter with 5 ml of hot 6 N hydrochloric acid solution (3.2) and twice with boiling water. Cool and make up to the mark with water. (The hydrochloric acid concentration of this solution should be about 0.5 N.)
Blank solution
6.2 Prepare a blank solution from which only the sample has been omitted and allow for this in the calculation of the final results.
Determination
6.3.1 Preparation of sample and blank test solutions
6.3.1Dilute the sample solutions (6.1.1 or 6.1.2) and the blank test solution (6.2), with 0.5 N hydrochloric acid solution (3.3) to a concentration within the optimal measuring range of the spectrophotometer. The final solution must contain 10% (V/V) of the lanthanum chloride solution (3.5).
6.3.2 Preparation of the calibration solutions
6.3.2By diluting the standard solution (3.4.2) with 0. 5 N hydrochloric acid solution (3.3) prepare at least 5 standard solutions of increasing concentration corresponding to the optimal measuring range of the spectrophotometer. The final solutions must contain 10% (V/V) of the lanthanum chloride solution (3.5).
Measurement
6.4 Set up the spectrophotometer (4.1), at a wave length of 279.5 nm using an oxidising air-acetylene flame. Spray successively, in triplicate, the standard solutions (6.3.2), the sample solution and the blank test solution (6.3.1), washing the instrument through with distilled water between each spraying. Plot the calibration curve using the mean absorbances as the ordinates and the corresponding concentrations of manganese υ/ml as the abscissae. Determine the concentration of manganese in the final sample and blank solutions by reference to the calibration curve.
EXPRESSION OF RESULTS
7. Calculate the manganese content of the sample taking into account the weight of the test sample and the dilutions carried out in the course of the analysis. Express the result either as a percentage or as mg/kg.
15.DETERMINATION OF THE NEUTRALISING VALUE IN LIMING MATERIALS
SCOPE AND FIELD OF APPLICATION
1. This method is applicable to products in Groups 5(a) and 5(b) of Section A of the Table in Schedule I of the Fertilisers Regulations 1990(41).
PRINCIPLE
2. The sample is dissolved in a measured quantity of standard hydrochloric acid, the excess of which is titrated with a standard solution of sodium hydroxide.
REAGENTS
3.—3.1 Hydrochloric acid, 0.5 N solution.
3.2 Sodium hydroxide, 0.5 N solution (carbonate free).
3.3 Phenolphthalein indicator solution: dissolve 0.25 g phenolphthalein in 150 ml 95% ethanol and dilute with water to 250 ml.
PREPARATION OF SAMPLE
4. Rapidly grind 50 g of the representative lime sample to pass through a 1 mm sieve.
PROCEDURE
Determination
5.—5.1 Weigh to the nearest 0.001 g, 0. 5 g of the prepared sample and transfer to a 300 ml conical flask. Add 50 ml of 0.5 N hydrochloric acid (3.1), cover the flask with a watch glass and boil the contents gently for five minutes. Cool the mixture to room temperature, add two or three drops of the phenolphthalein indicator (3.3) and titrate with 0.5 N sodium hydroxide solution (3.2) to the end point of the indicator.
EXPRESSION OF RESULTS
6. Determine the amount of hydrochloric acid consumed by the sample. 1 ml 0.5 N hydrochloric acid=0.01402 g calcium oxide (CaO).
The neutralising value is expressed as a percentage by weight of calcium oxide (CaO) and refers to undried sample as received.
16.DETERMINATION OF FINENESS OF PRODUCTS OTHER THAN POTASSIC BASIC SLAG
SCOPE AND FIELD OF APPLICATION
1. This method is applicable to “Rock phosphate” in Group 2(b) and to products in Groups 4(c), 5(a) and 5(b) of Section A of the Table in Schedule I of the Fertilisers Regulations 1990(42).
PRINCIPLE
2. By hand sieve shaking, the proportion of material passing through the prescribed sieve is determined.
APPARATUS
3. Sieves having square apertures of 45 mm, 6.7 mm, 6.3 mm, 5 mm, 3.35 mm, 1.0 mm and 150 microns; lower receiver to fit sieve. Test sieves conforming to British Standard 410: 1986 are suitable.
PROCEDURE
For sieving through 3.5 mm, 1.0 mm and 150 micron sieves
4.—4.1 Thoroughly mix the sample and quarter down until a portion of about 100 g is obtained. Heat this portion at 1OO°C until dry and thoroughly mix. Weigh to the nearest 0.01 g, 20 g and transfer to the sieve with the lower receiver attached. Proceed as described in 4.4.
For sieving through 6.7 mm, 6.3 mm and 5 mm sieves
4.2 Oven dry the sample at 1OO°C for 24 hours and thoroughly mix. Weigh to the nearest 0.1 g, 200 g and transfer to the sieve with the lower receiver attached. Proceed as described in 4.4.
For sieving through a 45 mm sieve
4.3 If the sample appears moist or damp, oven dry at 1OO°C for 24 hours, but if the sample appears dry, heating is not necessary. Thoroughly mix the sample and weigh to the nearest 0.1 g, 500 g and transfer to the sieve with the lower receiver attached. Proceed as in 4.4.
Sieving
4.4 Shake the sieve for 5 minutes, frequently tapping the side. Disintegrate soft lumps such as can be caused to crumble by the application of the fibres of a soft brush, taking care that the hard part of the brush does not make contact with the sieve and that the brush is not used to brush particles through the sieve. Brush out the powder in the lower receiver and weigh. Replace the receiver and repeat the shaking and tapping procedure for 2 minutes. Add the powder in the receiver to the first portion and weigh. Repeat the process until not more than 0.04 g passes through the seive during 2 minutes.
EXPRESSION OF RESULTS
5. Calculate the fineness by expressing the weight of the material passing through the sieve as a percentage of the weight of the portion of the dried (or as the case may be, undried) sample taken for sieving.
17.DETERMINATION OF FINENESS OF POTASSIC BASIC SLAG
SCOPE AND FIELD OF APPLICATION
1. Exclusively to “Potassic basic slag” in Group 3(b) of Section A of the Table in Schedule 1 of the Fertilisers Regulations 1990(43).
PRINCIPLE
2. By hand sieve shaking and dissolution of the soluble salts, the proportion of slag passing through the prescribed sieve is determined.
APPARATUS
3. Sieve having square apertures of 0.5 mm (500 microns); lower receiver to fit sieve. Test sieves conforming to British Standard 410: 1986 are suitable.
PROCEDURE
Preparation of the sample
4.—4.1 Thoroughly mix the sample and quarter down until a portion of about 100 g is obtained. Heat this portion at 1OO°C until dry and thoroughly mix.
Sieving
4.2 Weigh to the nearest 0.1 g, 20 g of the dry sample and transfer to the sieve with the lower receiver attached. Shake the sieve for five minutes, frequently tapping the sides. Disintegrate soft lumps that can be caused to crumble by the application of a soft brush, taking care that the hard part of the brush does not make contact with the sieve and that the brush is not used to brush particles through the sieve.
Transfer the finer portion from the container into a 500 ml beaker and add 200 mi of previously boiled water. Stir and then filter through a weighed glass sintered crucible. Thoroughly wash the residue with water, dry and re-weigh the crucible. Calculate the weight of slag in the mixture with a particle size of less than 0.5 mm (A).
Weigh to the nearest 0.01 g, about 20 g of the dry sample and transfer to a 500 ml conical flask. Add 200 ml previously boiled water and shake for 30 minutes. Filter through a weighed, sintered glass crucible, wash the residue thoroughly with water, dry and re-weigh the crucible. Calculate the total weight of slag in the mixture (B).
EXPRESSION OF RESULTS
5.

APPENDIX TO SCHEDULE 2



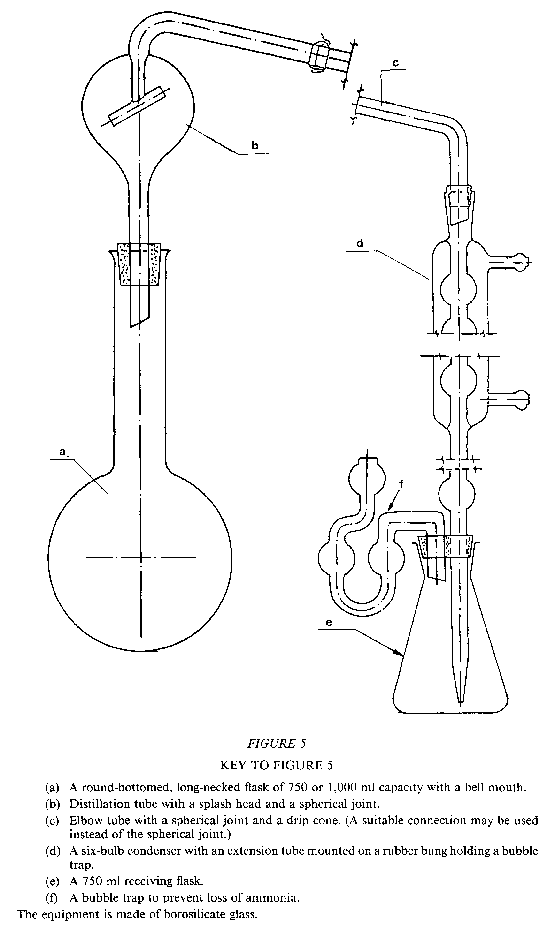



Whatman 42 or equivalent.
Whatman 42 or equivalent.
Whatman 541 or equivalent.
Whatman 541 or equivalent.
Whatman 541 or equivalent.
Commercially available standard copper solution may be used.
Whatman 541 or equivalent.
Commercially available standard iron solution may be used.
Whatman 541 or equivalent.
Whatman 541 or equivalent.
Commercially available standard manganese solution may be used.
Whatman 541 or equivalent.
Biuret can be prified beforehand by washing with ammoniacal solution (10%), then with acetone and drying in a vacuum.
Where the fertiliser is normal superphosphate or concentrated superphosphate in Group 2(a) of Section A, or NPK fertiliser in Group 1, NP fertiliser in Group 2, or PK fertiliser in Group 4 of Section B or NPK fertiliser suspension, NP fertiliser suspension or PK fertiliser suspension in Section C of the Table in Schedule 1 of the Fertilisers Regulations 1990.
Where the fertiliser is triple superphosphate in Group 2(a) of Section A, or NPK fertiliser containing soft ground rock phosphate or partially solubilised rock phosphate in Group 1, or NP fertiliser containing soft ground rock phosphate or partially solubilised rock phosphate in Group 2, or PK fertiliser containing soft ground rock phosphate or partially rock phosphate in Group 4 of Section B of the Table in Schedule 1 of the Fertilisers Regulations 1990.
If no mechnical shaker is available, the flask may be shaken by hand every 5 minutes.
Phosphorus soluble in mineral acids, water soluble phosphorus, phosphorus soluble in solutions of ammonium citrate, phosphorus soluble in 2% citric acid and phosphorus soluble in 2% formic acid.
21 ml when the solution to be precipitated contains more than 15 ml of citrate solution (neutral citrate, Petermann or Joulie alkaline citrate).
To precipitate phosphate solutions containing more than 15 ml citrate solution (neutral, Petermann or Joulie) which have been acified with 21 ml concentrated nitirc acid (see footnote to paragraph 6.1) use 80 ml of the precipitating reagent.
A reaction time of one-and-a-half hours is sufficient in the case of most of the organic substances in the presence of silver nitrate catalyst.
Commercially available standard copper solution may be used.
Whatman 541 or equivalent.